Day 1 :
Keynote Forum
Horst Köppel
University of Heidelberg, Germany
Keynote: Non adiabatic molecular dynamics following photoexcitation: an ab initio quantum approach
Time : 09:30-10:05

Biography:
Horst Köppel has been a Senior Lecturer, and later Professor of Theoretical Chemistry at Heidelberg University since 1991. His research interests focuses on the theory of the Jahn-Teller effect, molecular excited state processes and vibrational structure in electronic spectra; special focus is on phenomena which cannot be described within the conventional Born-Oppenheimer separation of electronic and nuclear motions. He has authored and coauthored about 250 peer-reviewed original research papers, edited three books with topical reviews and organized several conferences in the field.
Abstract:
The author will present an overview over the theoretical work on molecular dynamics following photoexcitation in the visible or UV spectral range. This carries the system to an electronically excited state with a potential energy surface (PES) differing from that of the electronic ground state. A rich variety of vibrational and related processes will thus be initiated which can proceed on a single or several of the many different excited-state PESs. If some of these PESs are close in energy they interact and the nuclear motion on them does not proceed independently. This is the realm of nonadiabatic molecular dynamics which has been in the focus of interest of spectroscopists and physical and theoretical chemists for many years. It is of fundamental importance for many different excited-state processes in biology, chemistry and physics (such as charge transfer, photochemical rearrangements etc.). A typical scenario are so-called conical intersections of potential energy surfaces where different PES become degenerate upon variation of two nuclear coordinates. Systems treated by us recently comprise SO2, small polyenes like butadiene or hexatriene or the benzene cation. Key features of the systems and methods used will be highlighted in the talk.

Schematic repres-entation of two conical inter-sections and their associated photochemical reaction pathways.
Keynote Forum
Pavel Hobza
Institute of Organic Chemistry and Biochemistry – CAS, Czech Republic
Keynote: Noncovalent interactions in bio- and nanosciences: quantum mechanical aproach
Time : 10:05-10:40

Biography:
Pavel Hobza (dr.hc, FRSC) obtained his PhD (1974) from the Institute of Physical Chemistry of the Academy of Science of the Czech Republic. He is a Distinguished Chair at the Institute of Organic Chemistry and Biochemistry of the Academy of Sciences of the Czech Republic, Prague, Czech Republic. His research team works on noncovalent interactions and their applications in bio- and material sciences, databases of accurate interaction energies and on in silico drug design. He has co-authored 4 books, more than 450 papers and 35 review papers in peer-reviewed journals, his H-index (WOS) is 98, sum of Times Cited is more than 35,000. He was awarded by “Highly Cited Researcher” in chemistry during 2014-2016 (Thomson Reuters, later Clarivate Analytics) and Schrödinger Medal in 2017 (World Association of Theoretical and Computational Chemists).
Abstract:
Noncovalent interactions play an important role in chemistry, physics and biology. Reliable characteristics like stabilization energy, structure and vibrational frequencies are obtained using composite coupled cluster schemes which offer the possibility of improving the accuracy of results obtained by adding excitation operators of increasing order. It was shown that already CCSD(T)/ CBS method yields an accurate and reliable description of noncovalent interactions, yet is only applicable to systems with several tens of atoms. Lower-level methods like DFT or semiempieical QM (SQM) should be parametriezed or verified and here the databases of accurate stabilization energies and geometries developed in our laboratory (S22, S66, X40 and L7) play an indispensable role. Binding free energy for host-guest and protein-ligand complexes is constructed as a sum of gas-phase interaction energy (ΔEint), change of desolvation free-energy (ΔΔGsolv), change of the conformational free energy of both components (ΔGconfw) and entropy term (eq. 1): ΔGw ≈ ΔEint + ΔΔGsolv + ΔGconfw – TΔS (1). Because of the size of systems investigated the DFT-D, and PM6 or SCC-DF-TB SQM methods combined with COSMO technique were considered. Performance of these methods was verified by comparison of interaction energies of model complexes with the benchmark values obtained from CCSD(T) and MP2.5 methods. Applicability of procedures described is demonstrated for evaluation of binding free energies of several extended systems like host – guest, protein – ligand and surface – admolecule ones.
- Theoretical and Computational Chemistry | Physical Chemistry: A Molecular Approach | Chemical Physics and Chemical Kinetics
Location: Macallan Glenfiddich

Chair
Masahiko Hada
Tokyo Metropolitan University, Japan

Co-Chair
Małgorzata A. Broda
University of Opole, Poland
Session Introduction
Masahiko Hada
Tokyo Metropolitan University, Japan
Title: Vanadium NMR chemical shift in vanadium complex catalyst: a cooperation of QC calculation and MLR analysis
Time : 10:55-11:20

Biography:
Masahiko Hada is a Professor at Tokyo Metropolitan University, Japan. Previously he worked as Associate Professor at Kyoto University, Japan from 1989-2002. He obtained his PhD from Kyoto University in the year 1986. He has his interests centered around quantum-chemical theories for accurate electronic states of molecules including electron correlation and relativistic effects, and computational approaches for analyses of chemical reactions, spectroscopies and molecular properties. Recently his expertise is extended to 2c-relativistic quantum-chemical theories and its applications to NMR, electronic excited states and spectroscopies (UV, CD, MCD, CPL), and chemical reactions on metal and metal-oxide surfaces, and chemical reaction mechanisms of enzyme containing transition metals.
Abstract:
The vanadium complex catalysts play an important role in olefin polymerization. (imido)vanadium(V) complexes containing (2-anilidomethyl)pyridine ligands were presented notable catalytic activity and high selectivity in ethylene dimerization. In recent studies, Nomura and his coworkers found a good relationship between the catalytic activity and the vanadium chemical shift (51V-NMR) for the (imido)vanadium complex in ethylene polymerization. They deduced that the high catalytic reactivity originates from stabilization of the active site by electron-donating substituents. The trend of observed 51V-NMR chemical shifts was well reproduced by the LC-BLYP/cc-pVTZ method as shown in Figure 1. Calculated 51V NMR chemical shifts were analyzed by the multiple linear regression analysis (MLRA) method with a series of calculated molecular properties as shown in Figure 2. We showed an accurate correlation (R2=0.95) between 51V-NMR chemical shifts and natural charge (Q), HOMO-LUMO energy gap (Δε), and Wiberg bond index of V=N bond (ω).
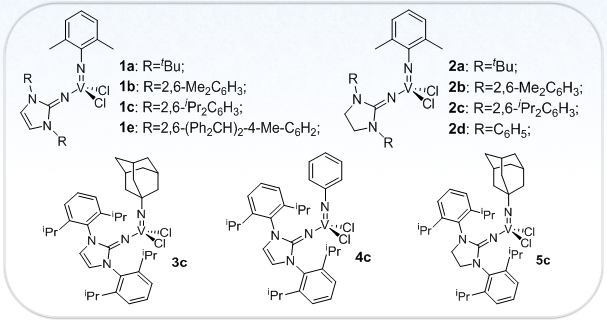
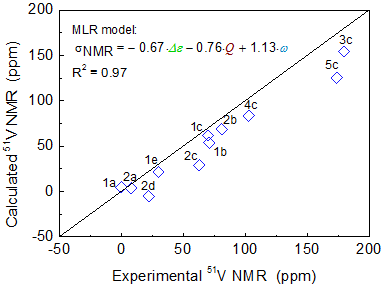
Figure 1. Molecular Structure of Vanadium Complexes [ref. 3]
Figure 2. MLR analysis of V-NMR chemical shifts
Dana Nachtigallová
Institute of Organic Chemistry and Biochemistry – CAS, Czech Republic
Title: Experimental and computational studies on the ground state of an isolated Fe(ii)-Phthalocyanine
Time : 11:20-11:45

Biography:
Dana Nachtigallová works as a senior researcher at the Institute of Organic Chemistry and Biochemistry of the CAS: Her current research interests focus on description of excited states of extended aromatic hydrocarbons and metallo-organic complex, simulations of nonadiabatic photodynamical processes, and modelling of noncovalent interactions of the excited state associates.
Abstract:
The electronic ground-state of the isolated Iron(II)Phthalocyanine (FePc) molecule is unknown. Previous experimental studies assigned the triplet as ground-state of FePc but they were performed either in gas phase at high temperature or in crystalline state. Using multi-reference CASPT2 and DMRG calculations we have shown that isolated FePc exhibits a quintet ground-state. This was further supported by MÓ§ssbauer spectroscopy and magnetic measurements of FePc in frozen non-polar chlorobenzene solvent which mimic the isolated condition of FePc.

Isa Degirmenci
Ondokuz Mayis University, Turkey
Title: Quantum chemical study on the tuning thiol-ene polymerization
Time : 11:45-12:10

Biography:
Isa Degirmenci completed his under-graduation from Marmara University in 1998-2002 and graduation from BoÄŸaziçi University in 2002-2005 respectively. He was awarded PhD from Gent University, Belgium and completed Post-doctoral studies from Australian National University. He has his expertise in structure-reactivity relationships in the free radical polymerization reactions. His investigations combine the understanding the structure properties of radical species and monomers with application of quantum chemical tools. His recent studies have simply explained the extraordinary reactivity and stability behavior of sulfur-centred radicals. These findings can be considered as paving the way for further utilizing recently emerged thiol-ene and thiol-yne polymerization reactions and also for further benefitting from self-healing reaction mechanisms of polymeric materials.
Abstract:
Recently, there has been a growing interest in the thiol-ene polymerization in many application areas from material science to bioorganic chemistry duo to the production of uniform polymer network, reduction of polymer shrinkage stress and obtaining narrow the Tg range. The most prominent feature of this polymerization is combination of the advantages of both step growth and chain growth polymerizations. The thiol-ene polymerization is involved in the initiation, propagation and chain transfer steps. This procedure is mostly governed by ratio of the propagation (kP) and chain transfer (kCT) reaction rates (kP/kCT). Recent experimental and theoretical studies have focused on ene functionality to evaluate the reaction procedure while the thiol functionality has been ruled out. However, this study has suggested that the thiol functionality also have to be taken into account to adjusting properties of a polymer. For this aspect, phenyl thiol derivatives have been considered for the thiol-ene polymerization of various monomers from electron deficient to electron rich alkenes. To evaluate kinetic and energetic features of the thiol-ene procedure, M06-2X/6- 31++G(d,p) level of theory was used for all geometry optimizations and frequency calculations. It was revealed that electrophilic nature of the phenylthio radicals and the stability of the RS-ene+ configuration predominates effect of the S-T gap of the ene functionality during addition reactions. Moreover, intermolecular interactions, such as π- π, have crucial role on the transition state of the chain transfer step. These interactions can diminish the strong influence of stability of intermediate carbon-centred radical on the chain transfer activation barrier. As a consequence, it was proved that the kP/kCT ratio is affected not only by ene functionality but also by the thiol functionality. This information can be taken into consideration for tailoring mechanical and physical properties of a polymer without changing the alkene structure to obtain industrially desirable polymer.
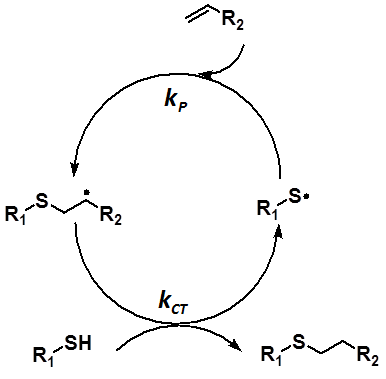
Scheme 1: Representation of the most significant steps of the thiol-ene reaction mechanism.
Laurentiu Spiridon
Institute of Biochemistry of the Romanian Academy, Romania
Title: Generalized coordinates hybrid Monte Carlo using high speed robotics algorithms
Time : 12:10-12:35

Biography:
Laurentiu Spiridon is currently working at the Institute of Biochemistry of the Romanian Academy, Romania. He has his work focused on creating a remote homology modelling method, SLIDE, a structural glycobiology software suite, Glyco Pack and a database of glycosylation sites named SAGS as. He is also working on optimizing the software package Robo Sampling which was developed during his Postdoctoral Fellowship (2013 - 2016) in Professor David Minh’s group at the Illinois Institute of Technology (IIT) in Chicago, USA. At IIT, he focused on developing enhanced sampling methods and for this software specifically - generalized coordinates Hamiltonian Monte Carlo using spatial operator algebra. His areas of interests include: force field development, more precisely he developed and implemented a flat-bottom harmonic potential that restricts ligands within the range of a specific pose during binding.
Abstract:
Simulations of large molecules, like proteins and DNA, are crucial for understanding chemical processes, relevant for medicinal and biochemical applications. As the information about macromolecular systems is accumulating from different types of experimental analysis the studied systems become larger and more complex – e. g. multimeric proteins - making their simulation ever more difficult. Even for simple systems, covering the conformational space while sampling from their proper Boltzmann distribution has proven challenging despite the recent increase in the computational power such as GPU (graphics processing unit) acceleration. One of the reasons for this drawback is that samplers that explore all the thermally accessible configuration space often become trapped in local minima. One way to overcome this is to use holonomic constraints on high-frequency degrees of freedom and group atoms into rigid bodies. One easy way to keep the constraints without imposing additional forces is to give up the Cartesian coordinates and encode the system in an alternate set of generalized coordinates such as BAT coordinates. However, when using arbitrary sets of generalized coordinates for low-frequency degrees of freedom solving the equations of motion requires the costly O(n3) inversion of the mass metric tensor. One solution is to use Jain spatial operator algebra (SOA) developed for robotics which allows to carry this inversion with O(n) complexity. Using recent generalizations of the equipartition principle, Fixman potential and Jain SOA in hybrid Monte Carlo trials to simulate molecular systems in generalized coordinates we were able to reproduce the Boltzmann distribution by drawing highly uncorrelated samples. Furthermore, by mixing fully flexible and random rigid body dynamics, we can achieve ergodicity by stratified sampling. The software needed for the abovementioned type of simulations is freely available online packed in a user-friendly easy to install package called Robo Sampling.
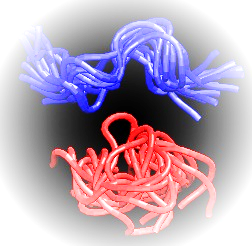
Figure 1. Regular MD (blue) vs GCHMC (red) simulations of a 9 amino acids tyrosinase peptide - YMD.
Tsveta Miteva
Sorbonne University, France
Title: Interatomic coulombic decay mediated by ultrafast superexchange energy transfer
Time : 12:35-13:00

Biography:
Tsveta Miteva received her bachelor’s and master’s degree in chemistry from Sofia University St. Kliment Ohridski, Bulgaria. She has her research focused on the theoretical description of electronic decay processes in atomic and molecular systems. In order to understand and model these ultrafast processes, she developed and implement original numerical tools. Another part of her research focuses on the electronic relaxation mechanisms following X-ray absorption in aqueous salt solutions.
Abstract:
Inner-valence ionized states of atoms and molecules live shorter if these species are embedded in an environment due to the possibility for ultrafast de-excitation known as interatomic Coulombic decay (ICD). In this process the initially excited species de-excites by transferring its energy to a neighbor and ionizing it within femtoseconds. The ICD lifetime, or going from the time to the energy domain, the ICD width, depends on the distance between the interacting monomers. At large interatomic separations, the process can be viewed as an exchange of a virtual photon between the monomers and thus, the decay width displays a 1/R6 dependence on the distance. In this work we show that the lifetime of the ICD active states decreases further when a bridge atom is in proximity to the two interacting monomers. This novel mechanism, termed superexchange ICD, is driven by the efficient transfer of excitation energy via virtual states of the bridge atom. As a showcase system we consider the NeHeNe trimer. The decay widths of the Ne2+(2s-1)2Σg+ state in the presence of He and in the isolated dimer were computed using the Fano- CI method. We demonstrate that the decay width of the Ne2+(2s-1)2Σg+ resonance increases 6 times in the presence of a He atom at a distance of 4 Å between the two Ne atoms. Using a simple model, we provide a qualitative explanation of the superexchange ICD and we derive an analytical expression for the dependence of the decay width on the distance between the neon atoms.
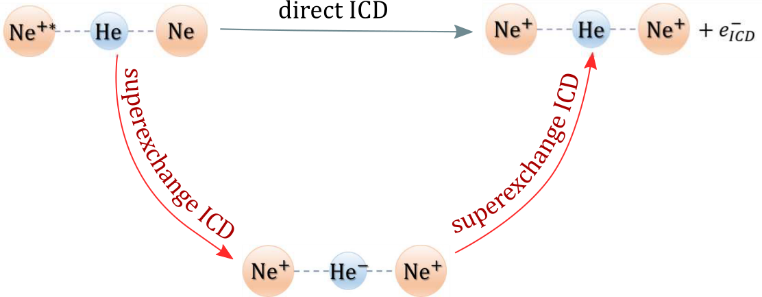
Figure 1. Schematic representation of the superexchange ICD process in NeHeNe trimer.
Kyung Yup Baek
Korea Advanced Institute of Science and Technology, Korea
Title: A smart strategy for finding global minima of microhydrated biomolecules with minimal DFT calculations
Time : 13:45-14:10

Biography:
Kyung Yup Baek has his research interests in identifying important physiochemical phenomena in vivo by modeling hydrated systems and understanding their conformationdependent properties. He obtained his Ph. D. in Chemistry from the Seoul National University in 2014 and is currently working as a postdoctoral researcher in the Korea Advanced Institute of Science and Technology (KAIST). Recently, he and his coworkers proposed an efficient computational methodology that can quickly and accurately find the most stable structures of hydrated biomolecular systems while drastically reducing the amount of DFT calculation that must be performed.
Abstract:
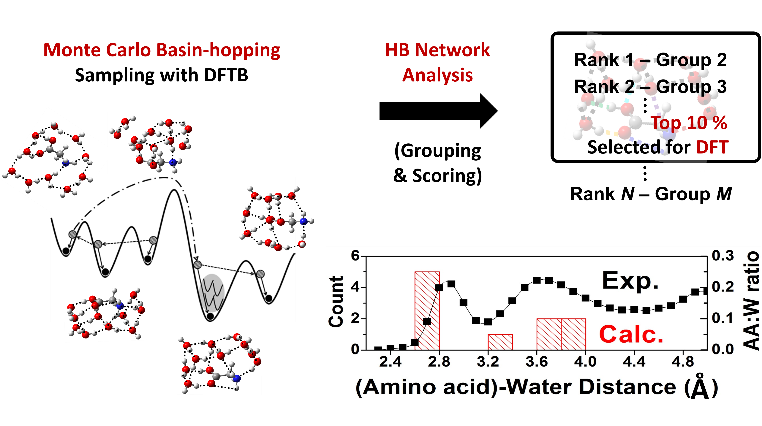
Water and biomolecules such as amino acids are the main building block of living organisms. Therefore, all phenomena and processes associated with them depend on the chemical interactions between these molecules in vivo. For example, it has been reported that tyrosine has a biological importance as a precursor of neurotransmitter called dopamine related to Parkinson's disease. Understanding their physicochemical behaviors in vivo is very challenging in terms of utilizing it for a new drug development. For this reason, biomolecule-water clusters as a model system have attracted significant attention in the scientific community. Undeniably, it is essential to identify stable structures and their patterns. However, there is no general way to elucidate stable hydrated structures even for simple amino acids because of the high complexity of chemical space increasing rapidly with the number of water molecules. In case of relying on chemical intuition only, it often failed to identify some important local minima, which could lead to misinterpretations about reaction dynamics of biomolecules in vivo. Here, we propose a very efficient computational method to selectively sample the most stable structures of the microhydrated biomolecules. The key idea is to utilize the unique structural patterns of H-bond networks obtained from their energetic features, i.e. their tendency to form more H-bonds. As a proof of concept, we could identify the new global minima of glycine•10(H2O) and for the first time, we found the minimum number of water molecules required to stabilize the zwitterionic form of tyrosine. Furthermore, the most stable structures of hydrated glycine and tyrosine indeed had common features, which were consistent with the X-ray data of proteins in water. Given the efficiency based on required DFT calculation amounts and accuracy, it is believed that our method give fast and accurate results for even more complex hydration systems.
Tadashi Ogitsu
Lawrence Livermore National Laboratory, USA
Title: Atomistic insights into electrochemical interfaces by ab-initio simulations and in-situ characterizations
Time : 14:10-14:35

Biography:
Tadashi Ogitsu was awarded PhD in Materials Science from University of Tsukuba, Japan in 1994 and completed his Post-doctoral studies from University of Illinois at Urbana-Champaign in 2001. He is a Deputy Group Leader of Quantum Simulation Group at the Lawrence Livermore National Laboratory and is the point of contact for DOE/EERE HydroGEN consortium which is designed to facilitate sustainable hydrogen production R&D. He has his expertise in ab-initio simulations and computational spectroscopy, and is interested in applying these skills and investigates on fundamental aspect of electrochemical processes relevant for energy applications such as photoelectrochemical hydrogen production.
Abstract:
Recent progresses in ab-initio computer simulation techniques as well as the advancement in high performance computing made direct simulations of complex systems such as electrochemical interfaces possible. Faithful modeling of electrochemical interface, however, is still very challenging partly due to lack of experimental probe that provides direct atomistic structural information. However, there are experimental characterization techniques that give us information about local chemical environment. In this presentation, we will first discuss about our recent experience in using ab-initio simulations for interpreting ambient-pressure X-ray photoemission spectroscopy (AP-XPS) results on III-V semiconductors (GaP/InP) exposed to chemical agents such as oxygen and/or water. XPS spectrum is usually analyzed based on comparison to reference information available in literature, which is valid if the system of our interest can be well approximated as a linear combination of well-defined reference problems, which is unlikely to be the case for electrochemical systems where local chemical environments tend to be dynamical in nature. Ab-initio simulations provide information regarding relation between thermodynamic stability of structural motifs and their spectroscopic signatures, therefore, peak assignment can be performed in a more rational and robust fashion. In addition, one may combine multiple theoretical and experimental spectroscopic information for the same system and may examine the consistency of each analysis. Development of accurate and realistic structural models of complex electrochemical systems will give us atomistic insights on electrochemical processes such as hydrogen/oxygen evolution and/or material corrosions, which in turn, can be used to improve the performances of energy conversion/storage devices.
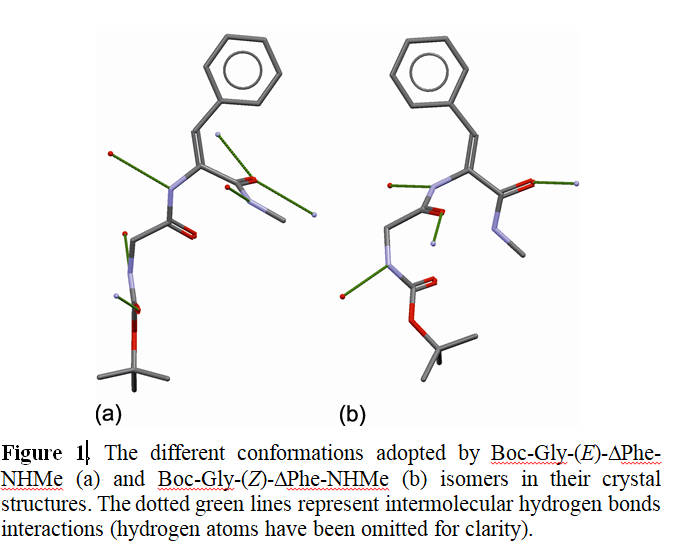
Małgorzata A. Broda
University of Opole, Poland
Title: Structure and spectroscopy of E and Z isomers of Boc-Gly-ΔPhe-NHMe
Time : 14:35-15:00

Biography:
Małgorzata A Broda is working as a Professor at Faculty of Chemistry, University of Opole, Poland. She has her expertise in molecular modeling and characterization of amino acid structure, electronic and spectroscopic properties. Her open minded approach to find energetic landscape of small, flexible biologically active molecules combines both theoretical prediction and experimental X-ray, NMR and IR studies. This results in wide interests about her research topic between other research groups and students. Her teaching of theoretical chemistry and spectroscopy at the University motivates several students to pursue the path of scientific carrier.
Abstract:
Biological activity of numerous small size molecules is directly related to their conformational properties. It is possible to control pharmaco-kinetic properties of naturally occurring peptides by introduction of nonstandard amino acid residues into their backbone chain which can result in analogues with improved pharmacological properties, such as resistance to enzymatic degradation, receptor selectivity, enhanced potency and bioavailability. For example, it is possible to introduce a dehydroamino acid residue and forcing a specific conformation of the chain fragment. Conformational properties of N-t-butoxycarbonylglycine-( E/Z)-dehydrophenylalanine N’-methylamides (Boc-Gly-(E/Z)- ΔPhe-NHMe) in chloroform were studied by NMR and IR spectroscopy. The low temperature crystal structure of the E isomer was determined by single crystal X-ray diffraction. The experimental findings were supported by extensive calculations at DFT (B3LYP, M06-2X) and MP2 levels of theory and the β-turn tendency for both isomers of the studied dipeptide were determined in vacuum and in solution. The obtained results reveal that the configuration of ΔPhe residue significantly affects the conformational properties of studied dehydropeptides. Theoretical conformational analysis reveals that the tendency to adopt β-turn conformations is much weaker for the E isomer (Boc-Gly-(E)- ΔPhe-NHMe), both in vacuum and in polar environment. We also showed a very good agreement between theoretical chemical shifts and calculated ones by using newly introduced relatively small and computationally inexpensive STO-3Gmag basis sets. The obtained results suggest a possibility of controlling dipeptide conformation by a simple chemical modification and thus allowing a future rational design of peptidomimetics.
Amrit Sarmah
Institute of Organic Chemistry and Biochemistry – CAS, Czech Republic
Title: Understanding the non-covalent interaction mediated modulations to the electronic structure of quasi-zero-dimensional graphene nanoflakes
Time : 15:00-15:25

Biography:
Amrit Sarmah pursued his PhD from the BITS Pilani, India. He is working in the field of theoretical and computational chemistry. After completing his PhD he had several international exposures in the field of theoretical chemistry and carried on his scientific endeavors at different places such as King Abdullah University of Science and Technology (KSA) Ben-Gurion University (Israel), JNCSR and IOCB (Czech Republic). His current research interest included computational design and fabrication of hybrid carbon nanomaterials and extensive theoretical investigation to tune their electronic and transport properties, modeling charge carrier mobility in 2D and quasi-2D semiconductors and non-covalent interaction mediated modulations to the spin-dependent properties of nanomaterials.
Abstract:
In the contemporary years, the magnetic or electric field induced modulations to the electronic environment of single molecular systems are a common practice. In this particular study, we have instigated the possibility of controlling the electronic and spin-dependent properties of hydrogen-terminated graphene fragments, so-called graphene nanoflakes (GNF) using weak noncovalent interaction as the external stimuli. The topological frustration in the graphene fragment appreciated the compelling electronic behavior of the system. This leads to some unorthodox spin-distribution in the system and it is possible to synchronize this electronic perturbation switching through a non-covalent interaction. These findings institute new avenue to sculpting such donor-acceptor composite as a self-regulated spintronic device in the next generation electronics.
Kazimierz Orzechowski
University of Wroclaw, Poland
Title: “Remake†of the non-linear dielectric effect in investigations of structure of liquids
Time : 15:40-16:05

Biography:
K Orzechowski specialized in physical and theoretical chemistry. He is currently a Professor at the University of Wroclaw, Poland. His research concerns physics and chemistry of dielectrics, phase transitions, intermolecular interactions, application of research in medicine, especially in cancer detection.
Abstract:
Structure of matter is determined by electrical interactions between molecules: dipole-dipole, dipole-ion, ion-ion interactions. Impact of an electric field on a matter allows to investigate these interactions and to understand the structure and dynamics of the investigated system. When a dielectric (in this case a liquid) is influenced by an external electric field, it undergoes polarization. For low-intensity field, the polarization is proportional to the field. However, if we increase the intensity of the electric field saturation effects could be expected. The measure of the so-called "non-linear dielectric effect" (NDE) is a nonlinear dielectric increment defined as the difference of the electric permittivity measured in a strong and in a weak electric field intensity: ΔεNDE = εE - εE->0. According to Debye classic theory, in liquids consisted of dipolar molecules, the increment should be negative and proportional to the square of the electric field. In many liquids, these requirements are fulfilled. Interestingly, deviations from the classical behaviors are sometimes observed. This happens when a strong external electric field affects conformation of molecules, disturbs association equilibria, significant deviations are also observed in the vicinity of phase transformations. NDE experiments were popular in the last decades of the 20th century. Measurements are difficult and commercial equipment for these studies is scarce. This may explain the recent decrease of interest in this particular technique. The report will present the NDE investigations of association phenomena in alcohols and critical phenomena in the vicinity of phase transformations. The details of the NDE experiment will be also presented. I firmly believe that a proper presentation of possibilities offered by a non-linear dielectric effect will cause an increase of interest in these studies and restore their rightful place in investigations of chemistry and physics of matter.
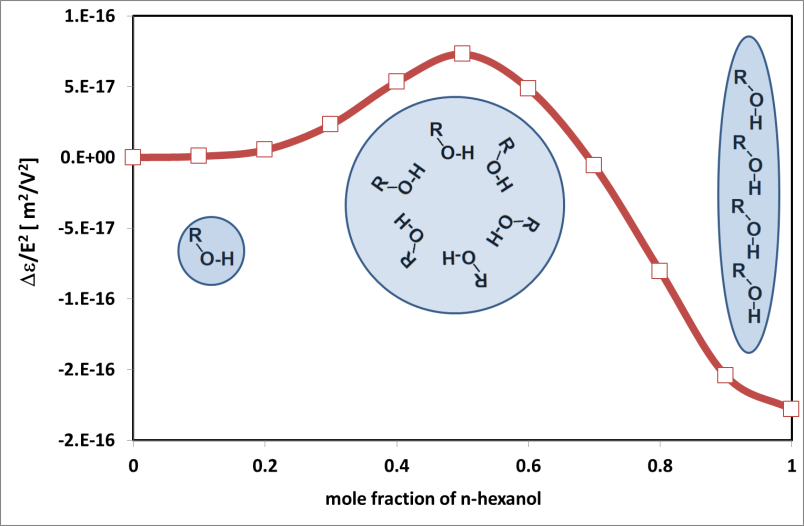
Fig. Piekara factor in n-hexanol-hexane mixture
Concha Tojo
University of Vigo, Spain
Title: Slowing-down of Pt reduction kinetics in microemulsions: a computer simulation study
Time : 16:05-16:30

Biography:
Concha Tojo has her expertise in computer simulation of chemical kinetics in compartmentalized media. Recently, her group is focused on the modelization of the synthesis of bimetallic nanoparticles in microemulsions. The aim of this research line is not only offer a better understanding of the complex mechanisms governing reactions in microemulsions, but open up a simple new way to synthesize bimetallic nanoparticles with ad-hoc controlled nanostructures.
Abstract:
The interest in developing new nanocatalysts composed by two metal components have been paid special attention, because the presence of a second metal gives rise to an improved catalytic behavior. In this line, Pt/M catalysts show high catalytic activity, which depends on the nanoparticle size, composition and metals segregation in the nanoparticle. A versatile method to control the size and composition of bimetallic nanoparticles is the micro emulsion route. But even here, the high number of variables make it difficult to tune the bimetallic nanostructure. As the ability to manipulate the metal distribution in bimetallic nanoparticles requires a deeper understanding of the mechanisms governing nanoparticle formation in microemulsions, we developed a simulation model to predict the atomic arrangement of bimetallic nanoparticles synthesized in microemulsions. The model was proved by comparing simulated nanostructures with Au/Pt nanoparticles. Excellent agreement between experimental and theoretical STEM profiles shows the validity of the model. On this basis, the model can be used not only as a tool to predict nanoparticles arrangement, but also as a means to further our knowledge of the kinetics in microemulsions. The purpose of this study is to perform a comprehensive kinetic analysis of the Pt/M nanostructures in the light of the interplay between three kinetic parameters: chemical reduction rates of the two metals, reactants concentration and inter micellar exchange rate. The combination of these factors determines the reaction rate of each metal, which in turn determines the final metal arrangement. We present results of Au/Pt (Pt: slower reduction) and Pt/Cu (Pt: faster reduction). Results show that Pt reduction rate not only depends on the usual parameters (Pt reduction rate, concentration, micro emulsion composition) but also on the reduction rate of another metal of the couple Pt/M. As a result, when Pt is the faster metal, final nanoparticle show a better metal segregation.
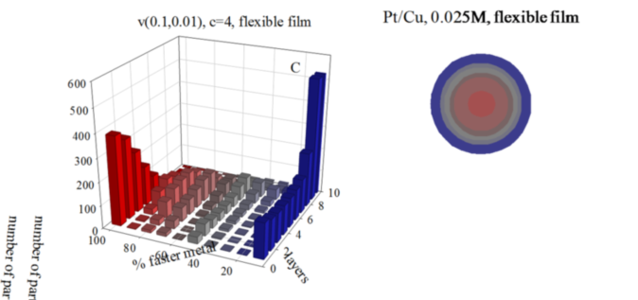
Figure: Histograms represent the number of particles with a given percentage of the faster metal in each layer, from the core to the surface, for different initial concentrations, and using a flexible film. Chemical rates: Au reduction is represented as an instantaneous reaction (100% of Au metal salts inside colliding micelles react); in Pt reduction only a 10% of Pt salts react in each collision; Cu, a 1% of Cu salts react. Scheme color: blue (0% - 45% of faster metal), grey (45%-55%), red (55% - 100%). Less red means less faster metal. Circles in each histogram represent nanoparticle structure in concentric layers, keeping the same color scheme
Janett Prehl
Chemnitz University of Technology, Germany
Title: Modeling reaction kinetics of twin polymerization via differential scanning calorimetry
Time : 16:30-16:55

Biography:
Janett Prehl has her expertise in theoretical physics with focus on modelling anomalous diffusion and complex systems. She got her Ph.D. in 2010 from the Technische Universität Chemnitz and works there as research assistant since 2009. In 2014 she got the possibility to work within the research unit FOR 1497 “Organic-Inorganic Nanocomposition through Twin Polymerization” financed by the German Research Foundation (DFG) with her own project “Simulation of structure formation, morphology and functionality of twin polymerization”. There she investigates the full structure formation process of twin polymerization on different length scales, from the atomic length scale via quantum chemical calculations and molecular dynamics simulations up to the mesoscopic length scale via Monte Carlo methods as the reactive bond fluctuation model.
Abstract:
The recently introduced method of twin polymerization is a synthesis route to produce nanoporous hybrid materials, containing organic and inorganic structure domains of 0.5 to 3 nm in a large variety of different compositions. Although first theoretical and experimental investigation has been performed, the open question still remains: How does the structure formation process of twin polymerization, yielding these interweaved organic-inorganic nanoporous hybrid materials, takes place in detail? Understanding the occurring effects and processes of the structure formation opens up the possibility to design (organic and/or inorganic) nanoporous materials with desired properties for industry. E.g. nanoporous materials are of great interest in applications like gas filter systems, catalyst or fuel cells. Here, we present a possibility to derive kinetic reaction parameters as the activation energy barrier or the reaction rate constant by fitting differential scanning calorimetry data via an equation system obtained from reaction kinetics.
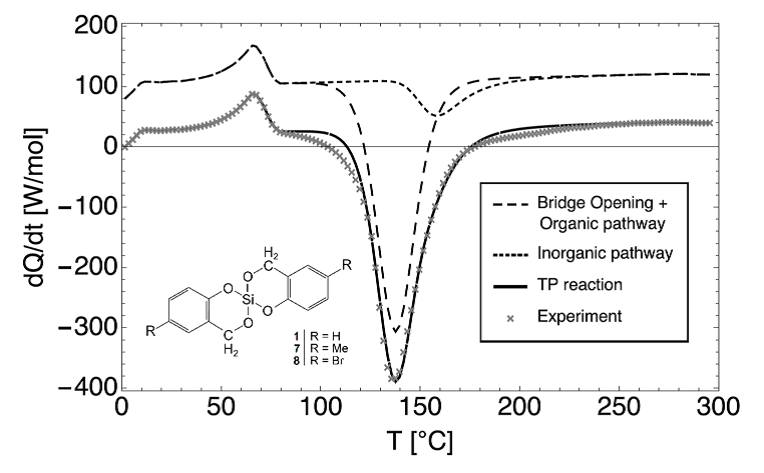
Figure 1: A possible reaction kinetics for the experimental DSC data represented via a two-step twin polymerization process.
Gregoire Guillon
University of Burgundy - Franche-Comté, France
Title: The O + O2 exchange reaction: symmetry, isotope effects, and influence of molecular forces
Time : 16:55-17:20

Biography:
Gregoire Guillon has his expertise in quantum scattering, inelastic and reactive, as well as in quantum Monte Carlo simulations. After working for several years in low temperature physics (cold molecules, helium droplets and hydrogen clusters), his latest results involve reactive processes occurring in an atmospheric chemistry context, as they are related to the ozone formation problem in stratosphere. He, together with PH, RC and VT has built this model after years of experience in research in laboratories worldwide.
Abstract:
Statement of the Problem: Molecular oxygen O2 is the most important molecule in Earth’s atmosphere and stratospheric ozone O3 protects us from 97% of UV radiations. The abundance in 16O being 99.8%, O2 and O3 exclusively formed from it are dominant, thereby giving a reference for any process involving oxygen. A strong enrichment (about 10%) of O3 in both 18O and 17O (the socalled mass-independent fractionation MIF), has first been observed decades ago. The three body recombination O + O2 + M → O3 + M is believed to be the main process leading to this enrichment and at low pressures, it can be partitioned into two steps: the formation of O3 in a highly excited rovibrational state, from reaction O + O2 → O3*, and its subsequent stabilization by collision with an energy absorbing partner M (say N2 or O2), O3* + M → O3 + M. Thus, the efficiency of the exchange reaction O + O2 → O3* → O2 + O, involving metastable O3* as an intermediate, is one of the key parameters to understand ozone formation. This reaction is very fast and competes with the stabilization process.
Methodology: Using a newly developed, very accurate, potential energy surface (PES), we have realized computationally intensive full-quantum investigation of the dynamics of this process, using a time-independent formalism.
Results: We have, from first principles, computed reactive cross sections and reproduced measured rate constant for the 18O + 32O2 process, within experimental error bars. We will sum up resulting cross sections and rate constants for the various 16O + 32O2, 18O + 32O2, 17O + 32O2, 16O + 36O2 and 16O + 34O2 processes, discussing isotope effects and inclusion of permutation symmetry. We will discuss the strong influence of the PES.