Day 2 :
Keynote Forum
Richard Tuckett
University of Birmingham, UK
Keynote: Climate change and global warming: thoughts of a quaker scientist
Time : 09:00-09:35

Biography:
Richard Tuckett completed his PhD in near-infrared spectroscopy in 1979. He first worked in electronic fluorescence spectroscopy of free radicals and molecular cations, often using supersonic beams and non-resonant electron excitation. From the late 1980s, he started using tunable vacuum-ultraviolet photon excitation from a synchrotron as a resonant ionisation source. In recent years he has studied the ionisation properties of long-lived greenhouse gases by threshold photoelectron and photoelectron photo-ion coincidence spectroscopy. Almost by accident, this has led him into atmospheric sciences and a wide interest in climate issues.
Abstract:
This talk arises from two articles recently accepted for publication by Elsevier in their Reference Modules [1,2]; the first also comes out next year in paper copy in the 3rd edition of Encyclopaedia Analytical Sciences, Written for the intelligent nonexpert, the science of the greenhouse effect and the most up-to-date data are presented in the first article [1]. In summary, the two most significant secondary greenhouse gases remain CO2 and CH4, together they contribute c. 80-85% of the secondary greenhouse effect, and this percentage has not changed for the last 20-30 years. CH4 could indeed prove to be as serious a secondary greenhouse gas as CO2. However, the total radiative forcing which causes the increase in Planet Earth’s temperature has increased consistently over this time window, and the huge majority of the world’s scientists now accept that we have a huge environmental issue on our hands that will not disappear. In the second article [2], suggestions are made as what issues people should think about from individual, government and world positions. The author is a practicing member of the Quaker (Society of Friends) religion, and throughout he comes to this problem from a moral viewpoint. This will not be a talk about religion, but rather how the six Quaker Testimonies (i.e. way we should lead our lives) on Truth and Integrity, Social Justice, Equality, Simplicity, Peace and Sustainability lead him in certain personal directions, and what advice he might give to Governments and World organisations (e.g. the United Nations). A concise and simple explanation of the Quaker religion in the UK in 2017 is written elsewhere [3]; much of it may surprise many delegates!.
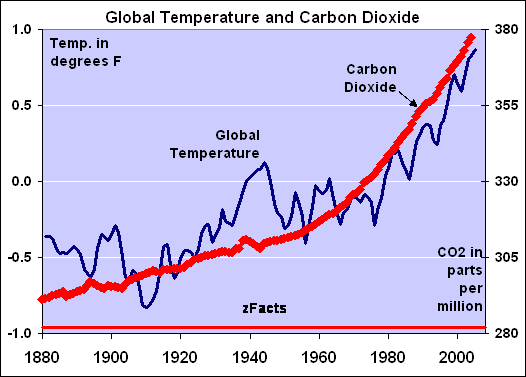
The average temperature of the Earth (red) and the concentration level of CO2 in the Earth’s atmosphere (in red) during the recent history since AD1880.
(Stoft http://zfacts.com/p/226.html or Hocker http://wattsupwiththat.com/2010/06/09/).
A rise of 1 F is equivalent to 0.56 oC. From a scientific viewpoint, there is no proven correlation between the two sets of data.
Keynote Forum
Alexander Lorenz
Paderborn University, Germany
Keynote: Manipulating liquid crystals via photo generated fields and tailored polymer
Time : 09:35-10:10

Biography:
Alexander Lorenz graduated from the Centre of Optoelectronics and Photonics Paderborn (Germany) in 2010 and has since conducted research at other leading institutions. He is the research Group Leader in the Department of Chemistry at the Paderborn University, Germany. His present research interests are photo generated polymerliquid crystal hybrids and inorganic-organic liquid crystal hybrids with high responsiveness and fast performance. He has completed Deutsche Forschungsgemeinschaft (DFG)- (a German research funding organization) -Research Fellowships in the Department of Engineering of the University of Cambridge UK and TU Berlin; led research projects funded by TU Berlin, DFG, and the US Air Force Office of Scientific research at TU Berlin and Paderborn University, and has (since 2017) acted as temporary replacement to fill the Full Chair Professorship for macromolecular chemistry and molecular materials at the Institute of Chemistry of the University of Kassel, Germany.
Abstract:
Liquid crystals (LCs) are well-known for their highly sensitive and tuneable optical properties. However, inorganic-organic hybrids with localized, light induced (opto-optical) responses and LC composites with fast or threshold-free switching are sought after. In addition to conventional modulation of the intensity, the main goal is to tune optical phase shifts of incident light waves. Localized optical responses can be triggered by the use of light, to allow for optical manipulation. Photo generated polymer can yield in highly responsive, fast LC composites for future displays and adaptive optics.
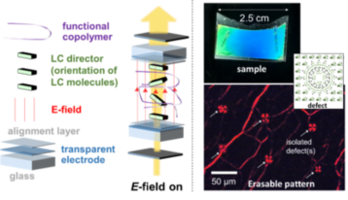
Figure 1: Schematic of a polymer network LC sample and polarized optical micrographs of a hybridized sample with photoinduced, erasable defect pattern.
Keynote Forum
Leonhard Grill
University of Graz, Austria
Keynote: Manipulation of single molecules at surfaces: switches, wires and motors
Time : 10:10-10:45

Biography:
Leonhard Grill is currently a Professor of Physical Chemistry at the University of Graz, Austria, since 2013. He studied physics at the University of Graz and did his PhD thesis at the Laboratorio TASC in Trieste (Italy) in experimental surface physics on electron scattering in ultrathin metal films (group of Silvio Modesti). He is an experimental physicist specialized in the study of single functional molecules. By using scanning probe microscopy, his group is able to image and manipulate individual atoms and molecules adsorbed at surfaces and to characterize specific molecular functions. In this way electronic, electrical, optical or mechanical properties of individual molecules are controlled with the goal to obtain fundamental physical and chemical understanding of these processes. He received the Feynman Prize in Nanotechnology (2011).
Abstract:
Molecular nanotechnology aims to use functional molecules as individual machines or electronic devices. Hence, their selfassembly into pre-defined architectures and the full control over each individual molecule are key objectives. Various examples of functional molecules, ranging from molecular wires to molecular switches and machines that are studied and manipulated at the single-molecule level by scanning tunneling microscopy (STM) under ultrahigh vacuum conditions, will be discussed in this presentation. Molecular wires or molecular nodes with different conjugation pathways can be fabricated from specifically designed molecular building blocks that are connected to two-dimensional networks or one-dimensional chains. In the case of molecular switches, the switching rate can be tuned up and down by only one single atom in the vicinity of the molecule. The same effect is then extended to molecular assemblies where cooperative effects in single molecules are directly observed. The switching process can also be used to trigger a molecular motor where the lateral translation of molecular machines on a surface can be enhanced by light of specific wavelengths that match the absorption properties of the molecule. By comparing molecules with and without a motor unit, the enhanced motion can be directly assigned to the motor that is incorporated in the molecules. STM manipulation gives detailed insight into the physical and chemical processes at the single-molecule level by varying the relevant parameters as tip height over the surface, bias voltage or tunneling current. While the speed is typically of minor importance in these experiments, it becomes crucial when studying so-called nanocars. By implementing a dipole moment into the molecular structure, we could show that very efficient and therefore fast manipulation can be realized. The key property is that no continuous imaging is required, rendering the manipulation fast enough to win the first nanocars race.
- Theoretical and Computational Chemistry | Physical Chemistry: A Molecular Approach | Chemical Physics and Chemical Kinetics
Location: Macallan Glenfiddich

Chair
Masahiko Hada
Tokyo Metropolitan University, Japan

Co-Chair
Małgorzata A. Broda
University of Opole, Poland
Session Introduction
Masahiko Hada
Tokyo Metropolitan University, Japan
Title: Vanadium NMR chemical shift in vanadium complex catalyst: a cooperation of QC calculation and MLR analysis
Time : 10:55-11:20

Biography:
Masahiko Hada is a Professor at Tokyo Metropolitan University, Japan. Previously he worked as Associate Professor at Kyoto University, Japan from 1989-2002. He obtained his PhD from Kyoto University in the year 1986. He has his interests centered around quantum-chemical theories for accurate electronic states of molecules including electron correlation and relativistic effects, and computational approaches for analyses of chemical reactions, spectroscopies and molecular properties. Recently his expertise is extended to 2c-relativistic quantum-chemical theories and its applications to NMR, electronic excited states and spectroscopies (UV, CD, MCD, CPL), and chemical reactions on metal and metal-oxide surfaces, and chemical reaction mechanisms of enzyme containing transition metals.
Abstract:
The vanadium complex catalysts play an important role in olefin polymerization. (imido)vanadium(V) complexes containing (2-anilidomethyl)pyridine ligands were presented notable catalytic activity and high selectivity in ethylene dimerization. In recent studies, Nomura and his coworkers found a good relationship between the catalytic activity and the vanadium chemical shift (51V-NMR) for the (imido)vanadium complex in ethylene polymerization. They deduced that the high catalytic reactivity originates from stabilization of the active site by electron-donating substituents. The trend of observed 51V-NMR chemical shifts was well reproduced by the LC-BLYP/cc-pVTZ method as shown in Figure 1. Calculated 51V NMR chemical shifts were analyzed by the multiple linear regression analysis (MLRA) method with a series of calculated molecular properties as shown in Figure 2. We showed an accurate correlation (R2=0.95) between 51V-NMR chemical shifts and natural charge (Q), HOMO-LUMO energy gap (Δε), and Wiberg bond index of V=N bond (ω).
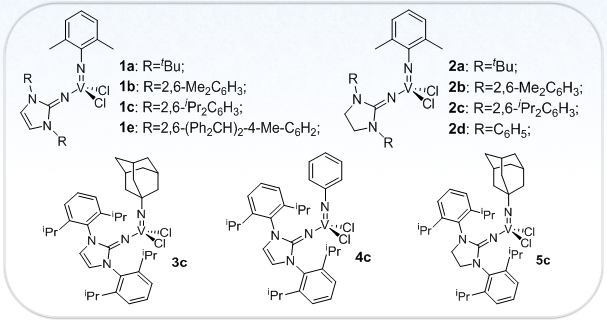
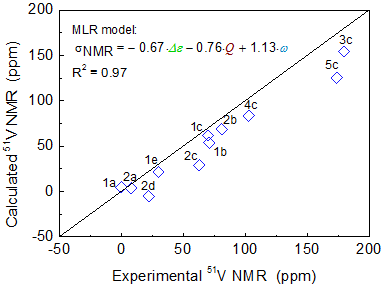
Figure 1. Molecular Structure of Vanadium Complexes [ref. 3]
Figure 2. MLR analysis of V-NMR chemical shifts
Dana Nachtigallová
Institute of Organic Chemistry and Biochemistry – CAS, Czech Republic
Title: Experimental and computational studies on the ground state of an isolated Fe(ii)-Phthalocyanine
Time : 11:20-11:45

Biography:
Dana Nachtigallová works as a senior researcher at the Institute of Organic Chemistry and Biochemistry of the CAS: Her current research interests focus on description of excited states of extended aromatic hydrocarbons and metallo-organic complex, simulations of nonadiabatic photodynamical processes, and modelling of noncovalent interactions of the excited state associates.
Abstract:
The electronic ground-state of the isolated Iron(II)Phthalocyanine (FePc) molecule is unknown. Previous experimental studies assigned the triplet as ground-state of FePc but they were performed either in gas phase at high temperature or in crystalline state. Using multi-reference CASPT2 and DMRG calculations we have shown that isolated FePc exhibits a quintet ground-state. This was further supported by MÓ§ssbauer spectroscopy and magnetic measurements of FePc in frozen non-polar chlorobenzene solvent which mimic the isolated condition of FePc.

Isa Degirmenci
Ondokuz Mayis University, Turkey
Title: Quantum chemical study on the tuning thiol-ene polymerization
Time : 11:45-12:10

Biography:
Isa Degirmenci completed his under-graduation from Marmara University in 1998-2002 and graduation from BoÄŸaziçi University in 2002-2005 respectively. He was awarded PhD from Gent University, Belgium and completed Post-doctoral studies from Australian National University. He has his expertise in structure-reactivity relationships in the free radical polymerization reactions. His investigations combine the understanding the structure properties of radical species and monomers with application of quantum chemical tools. His recent studies have simply explained the extraordinary reactivity and stability behavior of sulfur-centred radicals. These findings can be considered as paving the way for further utilizing recently emerged thiol-ene and thiol-yne polymerization reactions and also for further benefitting from self-healing reaction mechanisms of polymeric materials.
Abstract:
Recently, there has been a growing interest in the thiol-ene polymerization in many application areas from material science to bioorganic chemistry duo to the production of uniform polymer network, reduction of polymer shrinkage stress and obtaining narrow the Tg range. The most prominent feature of this polymerization is combination of the advantages of both step growth and chain growth polymerizations. The thiol-ene polymerization is involved in the initiation, propagation and chain transfer steps. This procedure is mostly governed by ratio of the propagation (kP) and chain transfer (kCT) reaction rates (kP/kCT). Recent experimental and theoretical studies have focused on ene functionality to evaluate the reaction procedure while the thiol functionality has been ruled out. However, this study has suggested that the thiol functionality also have to be taken into account to adjusting properties of a polymer. For this aspect, phenyl thiol derivatives have been considered for the thiol-ene polymerization of various monomers from electron deficient to electron rich alkenes. To evaluate kinetic and energetic features of the thiol-ene procedure, M06-2X/6- 31++G(d,p) level of theory was used for all geometry optimizations and frequency calculations. It was revealed that electrophilic nature of the phenylthio radicals and the stability of the RS-ene+ configuration predominates effect of the S-T gap of the ene functionality during addition reactions. Moreover, intermolecular interactions, such as π- π, have crucial role on the transition state of the chain transfer step. These interactions can diminish the strong influence of stability of intermediate carbon-centred radical on the chain transfer activation barrier. As a consequence, it was proved that the kP/kCT ratio is affected not only by ene functionality but also by the thiol functionality. This information can be taken into consideration for tailoring mechanical and physical properties of a polymer without changing the alkene structure to obtain industrially desirable polymer.
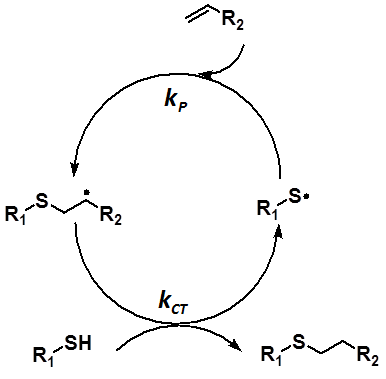
Scheme 1: Representation of the most significant steps of the thiol-ene reaction mechanism.
Laurentiu Spiridon
Institute of Biochemistry of the Romanian Academy, Romania
Title: Generalized coordinates hybrid Monte Carlo using high speed robotics algorithms
Time : 12:10-12:35

Biography:
Laurentiu Spiridon is currently working at the Institute of Biochemistry of the Romanian Academy, Romania. He has his work focused on creating a remote homology modelling method, SLIDE, a structural glycobiology software suite, Glyco Pack and a database of glycosylation sites named SAGS as. He is also working on optimizing the software package Robo Sampling which was developed during his Postdoctoral Fellowship (2013 - 2016) in Professor David Minh’s group at the Illinois Institute of Technology (IIT) in Chicago, USA. At IIT, he focused on developing enhanced sampling methods and for this software specifically - generalized coordinates Hamiltonian Monte Carlo using spatial operator algebra. His areas of interests include: force field development, more precisely he developed and implemented a flat-bottom harmonic potential that restricts ligands within the range of a specific pose during binding.
Abstract:
Simulations of large molecules, like proteins and DNA, are crucial for understanding chemical processes, relevant for medicinal and biochemical applications. As the information about macromolecular systems is accumulating from different types of experimental analysis the studied systems become larger and more complex – e. g. multimeric proteins - making their simulation ever more difficult. Even for simple systems, covering the conformational space while sampling from their proper Boltzmann distribution has proven challenging despite the recent increase in the computational power such as GPU (graphics processing unit) acceleration. One of the reasons for this drawback is that samplers that explore all the thermally accessible configuration space often become trapped in local minima. One way to overcome this is to use holonomic constraints on high-frequency degrees of freedom and group atoms into rigid bodies. One easy way to keep the constraints without imposing additional forces is to give up the Cartesian coordinates and encode the system in an alternate set of generalized coordinates such as BAT coordinates. However, when using arbitrary sets of generalized coordinates for low-frequency degrees of freedom solving the equations of motion requires the costly O(n3) inversion of the mass metric tensor. One solution is to use Jain spatial operator algebra (SOA) developed for robotics which allows to carry this inversion with O(n) complexity. Using recent generalizations of the equipartition principle, Fixman potential and Jain SOA in hybrid Monte Carlo trials to simulate molecular systems in generalized coordinates we were able to reproduce the Boltzmann distribution by drawing highly uncorrelated samples. Furthermore, by mixing fully flexible and random rigid body dynamics, we can achieve ergodicity by stratified sampling. The software needed for the abovementioned type of simulations is freely available online packed in a user-friendly easy to install package called Robo Sampling.
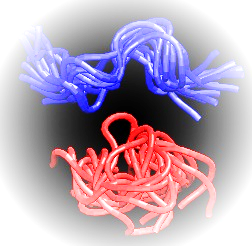
Figure 1. Regular MD (blue) vs GCHMC (red) simulations of a 9 amino acids tyrosinase peptide - YMD.
Tsveta Miteva
Sorbonne University, France
Title: Interatomic coulombic decay mediated by ultrafast superexchange energy transfer
Time : 12:35-13:00

Biography:
Tsveta Miteva received her bachelor’s and master’s degree in chemistry from Sofia University St. Kliment Ohridski, Bulgaria. She has her research focused on the theoretical description of electronic decay processes in atomic and molecular systems. In order to understand and model these ultrafast processes, she developed and implement original numerical tools. Another part of her research focuses on the electronic relaxation mechanisms following X-ray absorption in aqueous salt solutions.
Abstract:
Inner-valence ionized states of atoms and molecules live shorter if these species are embedded in an environment due to the possibility for ultrafast de-excitation known as interatomic Coulombic decay (ICD). In this process the initially excited species de-excites by transferring its energy to a neighbor and ionizing it within femtoseconds. The ICD lifetime, or going from the time to the energy domain, the ICD width, depends on the distance between the interacting monomers. At large interatomic separations, the process can be viewed as an exchange of a virtual photon between the monomers and thus, the decay width displays a 1/R6 dependence on the distance. In this work we show that the lifetime of the ICD active states decreases further when a bridge atom is in proximity to the two interacting monomers. This novel mechanism, termed superexchange ICD, is driven by the efficient transfer of excitation energy via virtual states of the bridge atom. As a showcase system we consider the NeHeNe trimer. The decay widths of the Ne2+(2s-1)2Σg+ state in the presence of He and in the isolated dimer were computed using the Fano- CI method. We demonstrate that the decay width of the Ne2+(2s-1)2Σg+ resonance increases 6 times in the presence of a He atom at a distance of 4 Å between the two Ne atoms. Using a simple model, we provide a qualitative explanation of the superexchange ICD and we derive an analytical expression for the dependence of the decay width on the distance between the neon atoms.
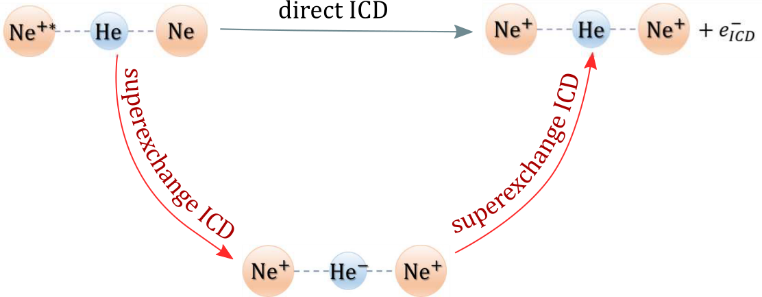
Figure 1. Schematic representation of the superexchange ICD process in NeHeNe trimer.
Kyung Yup Baek
Korea Advanced Institute of Science and Technology, Korea
Title: A smart strategy for finding global minima of microhydrated biomolecules with minimal DFT calculations
Time : 13:45-14:10

Biography:
Kyung Yup Baek has his research interests in identifying important physiochemical phenomena in vivo by modeling hydrated systems and understanding their conformationdependent properties. He obtained his Ph. D. in Chemistry from the Seoul National University in 2014 and is currently working as a postdoctoral researcher in the Korea Advanced Institute of Science and Technology (KAIST). Recently, he and his coworkers proposed an efficient computational methodology that can quickly and accurately find the most stable structures of hydrated biomolecular systems while drastically reducing the amount of DFT calculation that must be performed.
Abstract:
Water and biomolecules such as amino acids are the main building block of living organisms. Therefore, all phenomena and processes associated with them depend on the chemical interactions between these molecules in vivo. For example, it has been reported that tyrosine has a biological importance as a precursor of neurotransmitter called dopamine related to Parkinson's disease. Understanding their physicochemical behaviors in vivo is very challenging in terms of utilizing it for a new drug development. For this reason, biomolecule-water clusters as a model system have attracted significant attention in the scientific community. Undeniably, it is essential to identify stable structures and their patterns. However, there is no general way to elucidate stable hydrated structures even for simple amino acids because of the high complexity of chemical space increasing rapidly with the number of water molecules. In case of relying on chemical intuition only, it often failed to identify some important local minima, which could lead to misinterpretations about reaction dynamics of biomolecules in vivo. Here, we propose a very efficient computational method to selectively sample the most stable structures of the microhydrated biomolecules. The key idea is to utilize the unique structural patterns of H-bond networks obtained from their energetic features, i.e. their tendency to form more H-bonds. As a proof of concept, we could identify the new global minima of glycine•10(H2O) and for the first time, we found the minimum number of water molecules required to stabilize the zwitterionic form of tyrosine. Furthermore, the most stable structures of hydrated glycine and tyrosine indeed had common features, which were consistent with the X-ray data of proteins in water. Given the efficiency based on required DFT calculation amounts and accuracy, it is believed that our method give fast and accurate results for even more complex hydration systems.
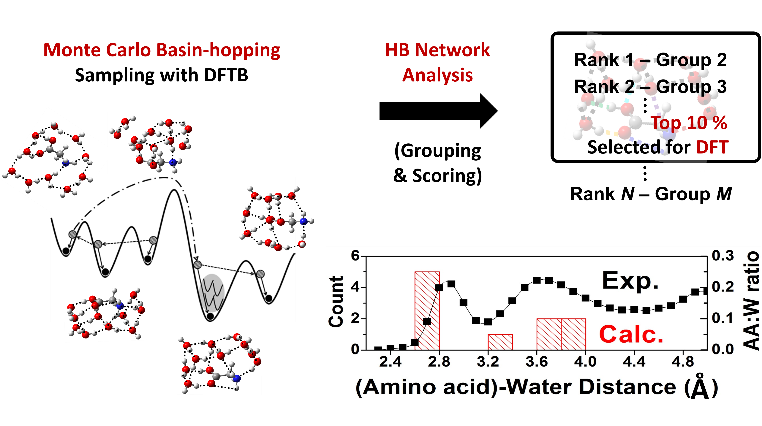
Tadashi Ogitsu
Lawrence Livermore National Laboratory, USA
Title: Atomistic insights into electrochemical interfaces by ab-initio simulations and in-situ characterizations
Time : 14:10-14:35

Biography:
Tadashi Ogitsu was awarded PhD in Materials Science from University of Tsukuba, Japan in 1994 and completed his Post-doctoral studies from University of Illinois at Urbana-Champaign in 2001. He is a Deputy Group Leader of Quantum Simulation Group at the Lawrence Livermore National Laboratory and is the point of contact for DOE/EERE HydroGEN consortium which is designed to facilitate sustainable hydrogen production R&D. He has his expertise in ab-initio simulations and computational spectroscopy, and is interested in applying these skills and investigates on fundamental aspect of electrochemical processes relevant for energy applications such as photoelectrochemical hydrogen production.
Abstract:
Recent progresses in ab-initio computer simulation techniques as well as the advancement in high performance computing made direct simulations of complex systems such as electrochemical interfaces possible. Faithful modeling of electrochemical interface, however, is still very challenging partly due to lack of experimental probe that provides direct atomistic structural information. However, there are experimental characterization techniques that give us information about local chemical environment. In this presentation, we will first discuss about our recent experience in using ab-initio simulations for interpreting ambient-pressure X-ray photoemission spectroscopy (AP-XPS) results on III-V semiconductors (GaP/InP) exposed to chemical agents such as oxygen and/or water. XPS spectrum is usually analyzed based on comparison to reference information available in literature, which is valid if the system of our interest can be well approximated as a linear combination of well-defined reference problems, which is unlikely to be the case for electrochemical systems where local chemical environments tend to be dynamical in nature. Ab-initio simulations provide information regarding relation between thermodynamic stability of structural motifs and their spectroscopic signatures, therefore, peak assignment can be performed in a more rational and robust fashion. In addition, one may combine multiple theoretical and experimental spectroscopic information for the same system and may examine the consistency of each analysis. Development of accurate and realistic structural models of complex electrochemical systems will give us atomistic insights on electrochemical processes such as hydrogen/oxygen evolution and/or material corrosions, which in turn, can be used to improve the performances of energy conversion/storage devices.
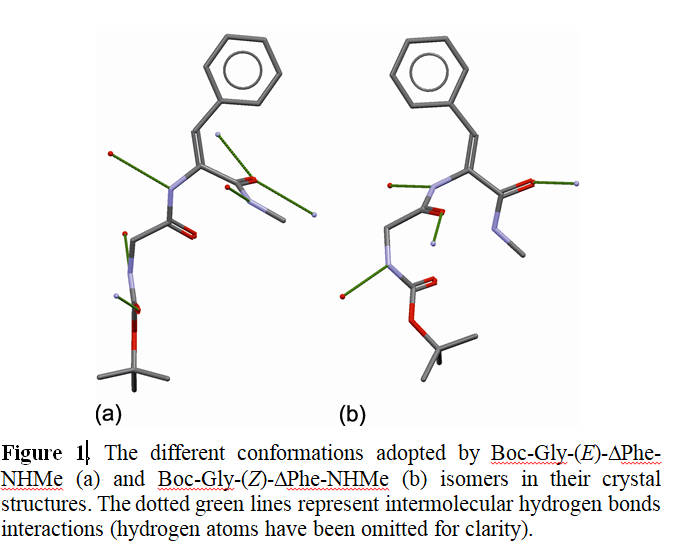
Małgorzata A. Broda
University of Opole, Poland
Title: Structure and spectroscopy of E and Z isomers of Boc-Gly-ΔPhe-NHMe
Time : 14:35-15:00

Biography:
Małgorzata A Broda is working as a Professor at Faculty of Chemistry, University of Opole, Poland. She has her expertise in molecular modeling and characterization of amino acid structure, electronic and spectroscopic properties. Her open minded approach to find energetic landscape of small, flexible biologically active molecules combines both theoretical prediction and experimental X-ray, NMR and IR studies. This results in wide interests about her research topic between other research groups and students. Her teaching of theoretical chemistry and spectroscopy at the University motivates several students to pursue the path of scientific carrier.
Abstract:
Biological activity of numerous small size molecules is directly related to their conformational properties. It is possible to control pharmaco-kinetic properties of naturally occurring peptides by introduction of nonstandard amino acid residues into their backbone chain which can result in analogues with improved pharmacological properties, such as resistance to enzymatic degradation, receptor selectivity, enhanced potency and bioavailability. For example, it is possible to introduce a dehydroamino acid residue and forcing a specific conformation of the chain fragment. Conformational properties of N-t-butoxycarbonylglycine-( E/Z)-dehydrophenylalanine N’-methylamides (Boc-Gly-(E/Z)- ΔPhe-NHMe) in chloroform were studied by NMR and IR spectroscopy. The low temperature crystal structure of the E isomer was determined by single crystal X-ray diffraction. The experimental findings were supported by extensive calculations at DFT (B3LYP, M06-2X) and MP2 levels of theory and the β-turn tendency for both isomers of the studied dipeptide were determined in vacuum and in solution. The obtained results reveal that the configuration of ΔPhe residue significantly affects the conformational properties of studied dehydropeptides. Theoretical conformational analysis reveals that the tendency to adopt β-turn conformations is much weaker for the E isomer (Boc-Gly-(E)- ΔPhe-NHMe), both in vacuum and in polar environment. We also showed a very good agreement between theoretical chemical shifts and calculated ones by using newly introduced relatively small and computationally inexpensive STO-3Gmag basis sets. The obtained results suggest a possibility of controlling dipeptide conformation by a simple chemical modification and thus allowing a future rational design of peptidomimetics.
Amrit Sarmah
Institute of Organic Chemistry and Biochemistry – CAS, Czech Republic
Title: Understanding the non-covalent interaction mediated modulations to the electronic structure of quasi-zero-dimensional graphene nanoflakes
Time : 15:00-15:25

Biography:
Amrit Sarmah pursued his PhD from the BITS Pilani, India. He is working in the field of theoretical and computational chemistry. After completing his PhD he had several international exposures in the field of theoretical chemistry and carried on his scientific endeavors at different places such as King Abdullah University of Science and Technology (KSA) Ben-Gurion University (Israel), JNCSR and IOCB (Czech Republic). His current research interest included computational design and fabrication of hybrid carbon nanomaterials and extensive theoretical investigation to tune their electronic and transport properties, modeling charge carrier mobility in 2D and quasi-2D semiconductors and non-covalent interaction mediated modulations to the spin-dependent properties of nanomaterials.
Abstract:
In the contemporary years, the magnetic or electric field induced modulations to the electronic environment of single molecular systems are a common practice. In this particular study, we have instigated the possibility of controlling the electronic and spin-dependent properties of hydrogen-terminated graphene fragments, so-called graphene nanoflakes (GNF) using weak noncovalent interaction as the external stimuli. The topological frustration in the graphene fragment appreciated the compelling electronic behavior of the system. This leads to some unorthodox spin-distribution in the system and it is possible to synchronize this electronic perturbation switching through a non-covalent interaction. These findings institute new avenue to sculpting such donor-acceptor composite as a self-regulated spintronic device in the next generation electronics.
Kazimierz Orzechowski
University of Wroclaw, Poland
Title: “Remake†of the non-linear dielectric effect in investigations of structure of liquids
Time : 15:40-16:05

Biography:
K Orzechowski specialized in physical and theoretical chemistry. He is currently a Professor at the University of Wroclaw, Poland. His research concerns physics and chemistry of dielectrics, phase transitions, intermolecular interactions, application of research in medicine, especially in cancer detection.
Abstract:
Structure of matter is determined by electrical interactions between molecules: dipole-dipole, dipole-ion, ion-ion interactions. Impact of an electric field on a matter allows to investigate these interactions and to understand the structure and dynamics of the investigated system. When a dielectric (in this case a liquid) is influenced by an external electric field, it undergoes polarization. For low-intensity field, the polarization is proportional to the field. However, if we increase the intensity of the electric field saturation effects could be expected. The measure of the so-called "non-linear dielectric effect" (NDE) is a nonlinear dielectric increment defined as the difference of the electric permittivity measured in a strong and in a weak electric field intensity: ΔεNDE = εE - εE->0. According to Debye classic theory, in liquids consisted of dipolar molecules, the increment should be negative and proportional to the square of the electric field. In many liquids, these requirements are fulfilled. Interestingly, deviations from the classical behaviors are sometimes observed. This happens when a strong external electric field affects conformation of molecules, disturbs association equilibria, significant deviations are also observed in the vicinity of phase transformations. NDE experiments were popular in the last decades of the 20th century. Measurements are difficult and commercial equipment for these studies is scarce. This may explain the recent decrease of interest in this particular technique. The report will present the NDE investigations of association phenomena in alcohols and critical phenomena in the vicinity of phase transformations. The details of the NDE experiment will be also presented. I firmly believe that a proper presentation of possibilities offered by a non-linear dielectric effect will cause an increase of interest in these studies and restore their rightful place in investigations of chemistry and physics of matter.
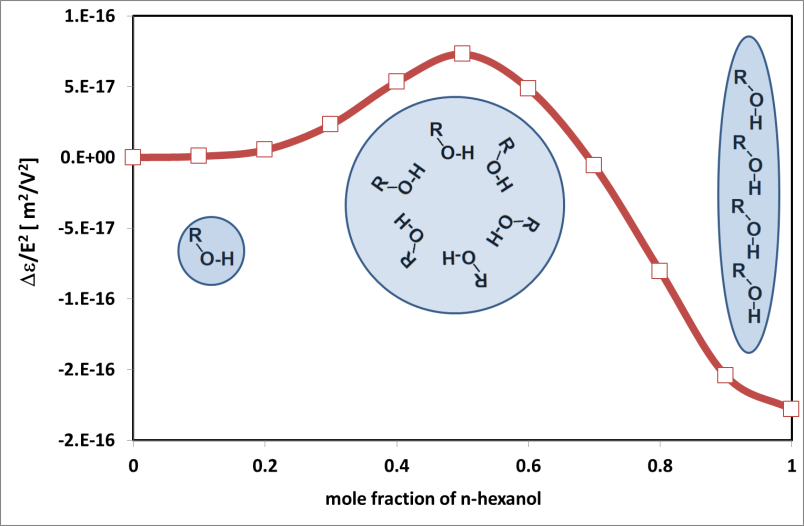
Fig. Piekara factor in n-hexanol-hexane mixture
Concha Tojo
University of Vigo, Spain
Title: Slowing-down of Pt reduction kinetics in microemulsions: a computer simulation study
Time : 16:05-16:30

Biography:
Concha Tojo has her expertise in computer simulation of chemical kinetics in compartmentalized media. Recently, her group is focused on the modelization of the synthesis of bimetallic nanoparticles in microemulsions. The aim of this research line is not only offer a better understanding of the complex mechanisms governing reactions in microemulsions, but open up a simple new way to synthesize bimetallic nanoparticles with ad-hoc controlled nanostructures.
Abstract:
The interest in developing new nanocatalysts composed by two metal components have been paid special attention, because the presence of a second metal gives rise to an improved catalytic behavior. In this line, Pt/M catalysts show high catalytic activity, which depends on the nanoparticle size, composition and metals segregation in the nanoparticle. A versatile method to control the size and composition of bimetallic nanoparticles is the micro emulsion route. But even here, the high number of variables make it difficult to tune the bimetallic nanostructure. As the ability to manipulate the metal distribution in bimetallic nanoparticles requires a deeper understanding of the mechanisms governing nanoparticle formation in microemulsions, we developed a simulation model to predict the atomic arrangement of bimetallic nanoparticles synthesized in microemulsions. The model was proved by comparing simulated nanostructures with Au/Pt nanoparticles. Excellent agreement between experimental and theoretical STEM profiles shows the validity of the model. On this basis, the model can be used not only as a tool to predict nanoparticles arrangement, but also as a means to further our knowledge of the kinetics in microemulsions. The purpose of this study is to perform a comprehensive kinetic analysis of the Pt/M nanostructures in the light of the interplay between three kinetic parameters: chemical reduction rates of the two metals, reactants concentration and inter micellar exchange rate. The combination of these factors determines the reaction rate of each metal, which in turn determines the final metal arrangement. We present results of Au/Pt (Pt: slower reduction) and Pt/Cu (Pt: faster reduction). Results show that Pt reduction rate not only depends on the usual parameters (Pt reduction rate, concentration, micro emulsion composition) but also on the reduction rate of another metal of the couple Pt/M. As a result, when Pt is the faster metal, final nanoparticle show a better metal segregation.
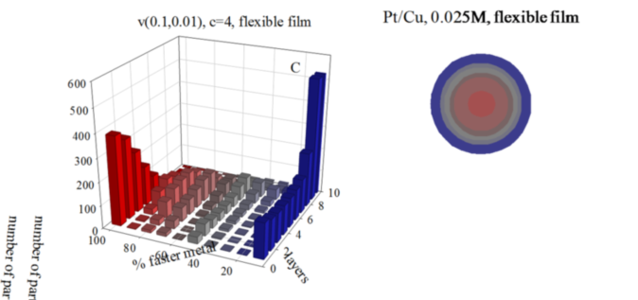
Figure: Histograms represent the number of particles with a given percentage of the faster metal in each layer, from the core to the surface, for different initial concentrations, and using a flexible film. Chemical rates: Au reduction is represented as an instantaneous reaction (100% of Au metal salts inside colliding micelles react); in Pt reduction only a 10% of Pt salts react in each collision; Cu, a 1% of Cu salts react. Scheme color: blue (0% - 45% of faster metal), grey (45%-55%), red (55% - 100%). Less red means less faster metal. Circles in each histogram represent nanoparticle structure in concentric layers, keeping the same color scheme
Janett Prehl
Chemnitz University of Technology, Germany
Title: Modeling reaction kinetics of twin polymerization via differential scanning calorimetry
Time : 16:30-16:55

Biography:
Janett Prehl has her expertise in theoretical physics with focus on modelling anomalous diffusion and complex systems. She got her Ph.D. in 2010 from the Technische Universität Chemnitz and works there as research assistant since 2009. In 2014 she got the possibility to work within the research unit FOR 1497 “Organic-Inorganic Nanocomposition through Twin Polymerization” financed by the German Research Foundation (DFG) with her own project “Simulation of structure formation, morphology and functionality of twin polymerization”. There she investigates the full structure formation process of twin polymerization on different length scales, from the atomic length scale via quantum chemical calculations and molecular dynamics simulations up to the mesoscopic length scale via Monte Carlo methods as the reactive bond fluctuation model.
Abstract:
The recently introduced method of twin polymerization is a synthesis route to produce nanoporous hybrid materials, containing organic and inorganic structure domains of 0.5 to 3 nm in a large variety of different compositions. Although first theoretical and experimental investigation has been performed, the open question still remains: How does the structure formation process of twin polymerization, yielding these interweaved organic-inorganic nanoporous hybrid materials, takes place in detail? Understanding the occurring effects and processes of the structure formation opens up the possibility to design (organic and/or inorganic) nanoporous materials with desired properties for industry. E.g. nanoporous materials are of great interest in applications like gas filter systems, catalyst or fuel cells. Here, we present a possibility to derive kinetic reaction parameters as the activation energy barrier or the reaction rate constant by fitting differential scanning calorimetry data via an equation system obtained from reaction kinetics.
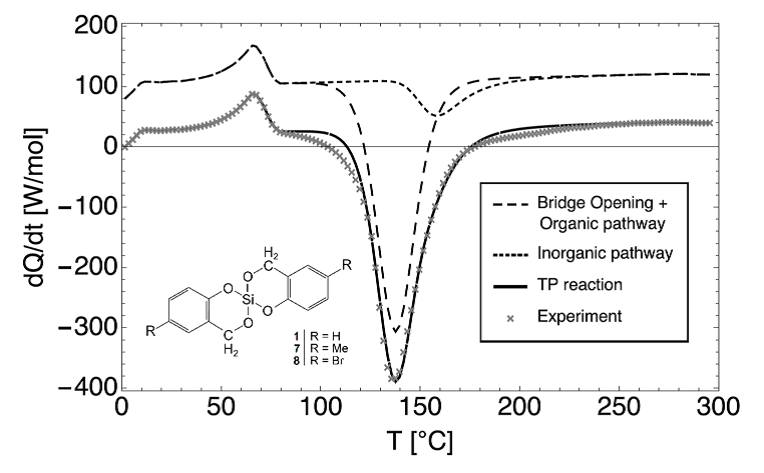
Figure 1: A possible reaction kinetics for the experimental DSC data represented via a two-step twin polymerization process.
Gregoire Guillon
University of Burgundy - Franche-Comté, France
Title: The O + O2 exchange reaction: symmetry, isotope effects, and influence of molecular forces
Time : 16:55-17:20

Biography:
Gregoire Guillon has his expertise in quantum scattering, inelastic and reactive, as well as in quantum Monte Carlo simulations. After working for several years in low temperature physics (cold molecules, helium droplets and hydrogen clusters), his latest results involve reactive processes occurring in an atmospheric chemistry context, as they are related to the ozone formation problem in stratosphere. He, together with PH, RC and VT has built this model after years of experience in research in laboratories worldwide.
Abstract:
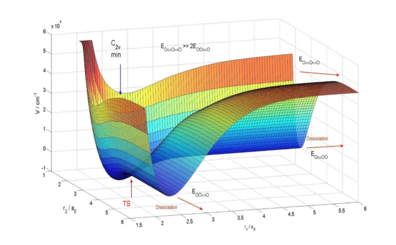
Statement of the Problem: Molecular oxygen O2 is the most important molecule in Earth’s atmosphere and stratospheric ozone O3 protects us from 97% of UV radiations. The abundance in 16O being 99.8%, O2 and O3 exclusively formed from it are dominant, thereby giving a reference for any process involving oxygen. A strong enrichment (about 10%) of O3 in both 18O and 17O (the socalled mass-independent fractionation MIF), has first been observed decades ago. The three body recombination O + O2 + M → O3 + M is believed to be the main process leading to this enrichment and at low pressures, it can be partitioned into two steps: the formation of O3 in a highly excited rovibrational state, from reaction O + O2 → O3*, and its subsequent stabilization by collision with an energy absorbing partner M (say N2 or O2), O3* + M → O3 + M. Thus, the efficiency of the exchange reaction O + O2 → O3* → O2 + O, involving metastable O3* as an intermediate, is one of the key parameters to understand ozone formation. This reaction is very fast and competes with the stabilization process.
Methodology: Using a newly developed, very accurate, potential energy surface (PES), we have realized computationally intensive full-quantum investigation of the dynamics of this process, using a time-independent formalism.
Results: We have, from first principles, computed reactive cross sections and reproduced measured rate constant for the 18O + 32O2 process, within experimental error bars. We will sum up resulting cross sections and rate constants for the various 16O + 32O2, 18O + 32O2, 17O + 32O2, 16O + 36O2 and 16O + 34O2 processes, discussing isotope effects and inclusion of permutation symmetry. We will discuss the strong influence of the PES.
- Solid-state Chemistry | Quantum Chemistry | Electro & Photochemistry | Physical Chemistry of Macromolecules
Location: Macallan Glenfiddich

Chair
Alexander Lorenz
Paderborn University, Germany

Co-Chair
Kazimierz Orzechowski
University of Wroclaw, Poland
Session Introduction
Vlasta BonaÄić-Koutecký
Humboldt University of Berlin, Germany
Title: Tuning optical and catalytic properties of ligated noble metal clusters by synergistic role of metallic and organic subunits
Time : 11:00-11:25

Biography:
Vlasta BonaÄić-Koutecký is a Professor in a Department of Chemistry, Humboldt University, Berlin. Since 2010 she has established the Interdisciplinary Center for Advanced Science and Technology (ICAST) at the University of Split, Croatia and became a head of Center of Excellence STIM in 2014. In the field of nanoscience Vlasta BonaÄić-Koutecký has recognized, before others, that metal nanoclusters (with only few atoms) have unique structural, optical and reactivity properties which are combining molecular-like with metallic features. This added a new unexpected dimension to the traditional nanoscience, introducing small metal nanoclusters into material science within the field of nanocatalysis for renewable energy as well as nanooptics and nano-biosensing for medical diagnostics.
Abstract:
Theoretical investigation of the linear and nonlinear optical properties of thiolate-protected low nuclearity noble metal clusters will be first presented. In this context theoretical approaches for reliable description of two-photon absorption spectra will be addressed. Goal is to design species exhibiting strong one-photon and/or two-photon absorption and emission in the UV-VIS spectral range. We will show that the optical properties can be tuned by creating the appropriate interplay between electronic excitations within the cluster core and selected prototype of ligands. Comparison with available experimental results will be discussed. We conclude that such low nuclearity protected noble metal clusters are promising for bio-labelling and imaging as alternatives to the standard fluorescent probes such as quantum dots or organic dyes. Second, we present our study of small coinage metal hydride ligated nanoclusters showing their capability to release the hydrogen. We propose the concept of synergistic role of ligand and substrate in catalysis on example of formic acid. This new catalyst neatly fits into a zeolite which does not perturb reactivity, thus providing a unique example on how “heterogenization” of a homogenous catalyst for the selective catalyzed extrusion of carbon dioxide from formic acid can be achieved, with important application in hydrogen storage and in situ generation of H2. The above results motivated us to investigate the selective decomposition of formic acid driven by highly porous aluminum based metal-organic framework in order to design new materials for the heterogenous catalysis. Thus, we illustrated that unique optical and reactivity properties of ligated noble metal clusters which can be tuned by appropriate interplay between metallic and organic subunits have significant potential for different applications.
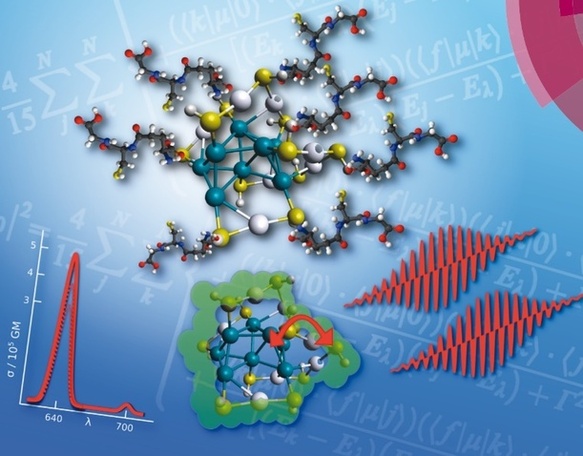
Figure 1: Two-photon absorption of ligand-protected Ag15 nanoclusters. Towards a new class of nonlinear optics nanomaterials.
Eugene B. Gordon
Russian Academy of Sciences, Russia
Title: Non-isothermal physico-chemical processes in superfluid helium
Time : 11:25-11:50

Biography:
Eugene B Gordon pursued his PhD in 1970 from Moscow University for Physics and Technology (MUPT) and Doctor of Science Degree in 1981 from the Institute of Problems of Chemical Physics (IPCP) of the Russian Academy of Sciences, Russia. He is currently the Principal Scientist of IPCP and Professor of Chemical Physics in MUPT. He has published more than 180 papers in reputed journals. He is the Member of All-Russia Supreme Qualification Committee; Member of Dissertation Councils in the IPCP and in Joint Institute of High Temperatures. He has his expertise in chemical kinetics, spectroscopy, chemical physics at low temperature.
Abstract:
Liquid helium cooled below T=2.17 K, namely superfluid helium, is homogeneous structureless liquid with ultrahigh thermal conductivity and it seemingly represents no interest as a matrix for chemical reactions. Indeed the reaction kinetics should be diffusion-controlled, and every collision at such low T would lead to instantaneous coalescence into loose fractal structure. These trivial arguments, fortunately, turned out to be fundamentally wrong. The point is that practically any perturbation of superfluid helium leads to the nucleation of quantized vortices, 1D (diameter ≈ 1Å, length≈ 1cm) excitations, which attract by Bernoulli forces any guest particle to vortex core. The captured particles freely moving towards each other along the core have much more probability to collide than in bulk liquid. As a result, the coagulation in superfluid helium proceeds mainly in vortices and leads to the formation of long thin nanowires, not spheres as usual. The ultrafast heat transfer in superfluid helium is associated with the laminar flow of its normal component. However this flow is turbulized already at modest values of the heat flux density equal to several W/cm2. In order to avoid self-fusion under two nanoclusters merging, the heat fluxes by orders of magnitude larger are necessary. As a result, liquid helium evaporates around the coagulation product forming a gas bubble of low pressure, which prevents the heat leakage. Thus, the product heats up to thousands of Kelvin and then melts, acquiring a spherical shape and a dense structure due to surface tension. And only provided the clusters grow up to certain size they become to stick together into nanowires. By using these effects we: (i) created the universal method for production of thin nanowires with perfect shape and structure, and (ii) realized in the laboratory the imitation of interstellar dust growth in the space.
Mouna Ben Yahia
ICGM, University of Montpellier, France
Title: Versatile approach combining theoretical and experimental aspects of raman spectroscopy to investigate battery materials
Time : 11:50-12:15

Biography:
Mouna Ben Yahia is an Assistant Professor at University of Montpellier, France. She is working on structural, mechanical, thermodynamic and vibrational properties of electrode materials for Li-ion batteries. She is developing a rapid and efficient characterization method based Raman spectroscopy for understanding the electrochemical mechanisms that occur in positive electrode materials within the lithium-ion batteries.
Abstract:
The Li-ion batteries are the most efficient devices in term of energy storage. The spinel LiNi0.5Mn1.5O4 (LNMO) is a promising positive electrode for lithium-ion batteries (LIBs) thanks to its high energy density and high voltage. Two LNMO polymorphs whose structural stabilities strongly depend on their synthesis conditions have been reported: ordered LNMO (P4332) and disordered LNMO (Fd-3m) on Ni/Mn atomic sites. Unfortunately, conventional X-ray diffraction cannot easily differentiate them. An easy and efficient way to do that is to use Raman scattering. Nevertheless difficulties were encountered to properly assign the observed vibration modes. Disordered LNMO is a typical case for which different approaches were used in the literature and conclusions were drawn based on only assumptions. Some people postulate for a discernible, other no-discernable Ni-O and Mn-O vibration bond in the Raman spectrum with no real proof to support their approach. The relatively new feature of modeling the Raman intensity in periodic system within DFT codes, allow us to resolve the last bottleneck of understanding the vibrational properties of spinel LNMO. For a given normal mode, the rationalization of the origin of the Raman intensities was done through a pertinent choice of descriptor resulting from a fine analysis of electronic structure. With this approach we assign all the normal modes and prove for the first time that the most intense peaks are mainly correlated to the Li-O contrary to what was reported in the literature. Also we confirm the assumption of discernible Ni-O and Mn-O vibration bonds. All these results will be discussed, to demonstrate that Raman spectroscopy coupled to calculated Raman intensities is a tool of choice to investigate cathode material for Li-ion batteries and more generally to follow the reaction mechanisms and possible intermediate species during electrochemical process.
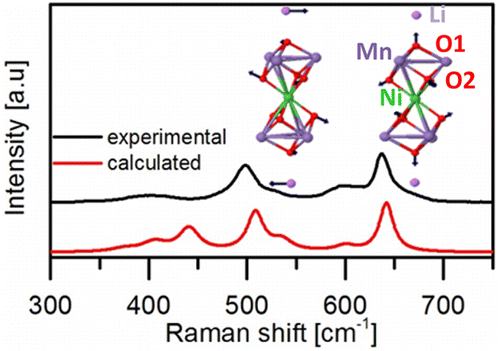
Experimental and calculated Raman spectra of the LNMO. Vibration modes of the most intense peaks.
Magdalena Sałdyka
University of Wrocław, Poland
Title: Photochemistry of acetohydroxamic acid in solid argon. FTIR and theoretical studies
Time : 12:15-12:40

Biography:
Magdalena SaÅ‚dyka obtained her PhD from the University of Wroclaw (Poland) under the supervision of Professor Zofia Mielke. She is continuing her work as an Assistant Professor at the same university Her research interests include: conformational analysis, photochemistry, inter- and intramolecular interactions – particularly in hydrogen bonded systems and theoretical modelling of molecular structure and vibrational spectra. She is an expert in low temperature matrix isolation technique combined with infrared spectroscopy. In the last years she has been developing studies on structural and spectroscopic properties of simple hydroxamic acids. Her academic teaching experience is related with methods of physicochemical analysis.
Abstract:
Statement of the Problem: Hydroxamic acids exhibit a wide spectrum of biological activities that stimulated progress in the chemistry of this class of compounds. They are known to be involved in iron transport phenomena and are active as antibiotics, antitumor and antifungal agents, and specific enzyme inhibitors. Extensive work has been carried out on the formation of hydroxamic acids, their reactions and structure in the ground state. However, the photochemical properties of hydroxamic acids are still not well recognized.
Methodology & Theoretical Orientation: The acetohydroxamic acid (AHA)/Ar matrices, prepared by co-deposition of AHA vapor coming out of the oven assembled inside the cryostat with a large excess of argon onto the cold CsI window, were exposed to 225 nm OPO radiation and to full output of the Xe lamp. The experimental studies were supported by ab initio calculations at the MP2/aug-cc-pVTZ level of theory.
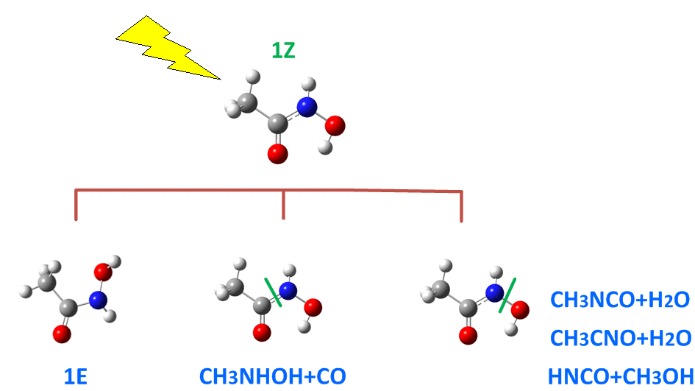
Findings: The performed irradiation of acetohydroxamic acid isolated in Ar matrices promotes the isomerization, 1Z → 1E, and AHA photodissociation reactions. Four pairs of coproducts are experimentally found in the photolysis, they form the complexes: CH3OH⋅⋅⋅HNCO (1), H2O⋅⋅⋅CH3NCO (2), H2O⋅⋅⋅CH3CNO (3) and CO⋅⋅⋅CH3NHOH (4). The comparison of the theoretical spectra with the experimental ones allowed to determine the structures of the complexes formed in the matrix.
Conclusion & Significance: The importance of the AHA molecule for biological and pharmaceutical applications triggers questions about the influence of UV-VIS irradiation on the photochemical properties of AHA. The mechanisms of the photodecomposition reaction channels leading to formation of the four co-products are proposed. It is concluded that the first step in formation of the (1), (2) and (3) complexes is the scission of the N-O bond, whereas the creation of the complex (4) is due to the cleavage of the C-N bond.
Valérie Brenner
Laboratory Interactions, Dynamics and Lasers-CEA Saclay, France
Title: Excited states deactivation in model proteins chains: nonadiabatic dynamics simulations and ab initio methods
Time : 12:40-13:05

Biography:
Valérie Brenner is a theoretical chemical physicist specialized in non-covalent interactions modeling and quantum chemistry computations of medium-sized and large molecular systems. She has always been working closely with different experimental teams and has acquired a wide experience in studying the physical chemistry of medium-sized and large molecular systems. After a period devoted to both modeling of intermolecular interactions and exploration of the large molecular systems potential energy surfaces during which she developed “homemade” codes, she has been recently invested in a new research field, i.e. computation of excited states of large molecular systems, focusing on the mechanisms of non-radiative relaxation in model protein chains.
Abstract:
Following UV absorption, many biomolecular systems are endowed with mechanisms of excited-states deactivation that ensure their photochemical stability. One of the major goals of our research is to investigate conformer-selective dynamics of biologically relevant molecular systems by an original innovative computational strategy in order to document the basic physical phenomena controlling the lifetime of excited states, highlighting the link between electronic dynamics and structure. This innovative multi-step computational strategy allows to both characterize the first excited states of bio-relevant systems and model efficiently their potential energy surfaces, using, first, nonadiabatic dynamics simulations based on time-dependent density functional theory (NA-TDDFT) to provide hints about the critical motions that drive the deactivation, which will then be investigated at a better level with two families of methods: i) the standard approximate coupled cluster singles and doubles method (CC2) and ii) and multireference (MR) methods. Developed on small capped peptide models and always backed up by key conformation selective gas phase experiments carried out in our team at several timescales,1,2 this innovative strategy is now applied to monohydrated capped peptides as well as capped dipeptides. We will present here the last results obtained on these systems. In addition, benchmark of the CC2 method on a set of model peptide chains (structure, energetic and vibrational frequencies of the first ππ* exited state)3,4 as well as assessment of the CC2 method validity from comparison with MR methods5 will be also reported.
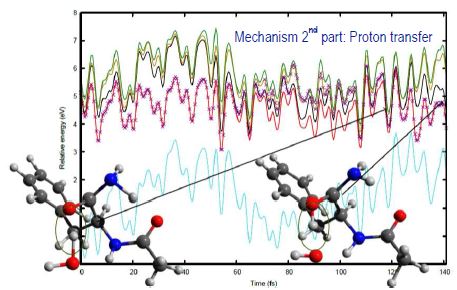
Figure: Illustration of a deactivation mechanism: Time dependence of the potential energy of the ground (blue) and four lowest excited states of NAPA-H2O along a selected nonadiabatic trajectory.
Yeshayahu Ben-Eliyahu
Ben-Gurion University of the Negev, Israel
Title: Computational and empiric considerations regarding the electrocatalytic reduction of CO2 by water soluble cobalt porphyrins
Time : 14:20-14:45

Biography:
Y Ben Eliyahu pursued all his degrees in Chemistry from the Hebrew University of Jerusalem, Jerusalem, Israel. His Msc and PhD theses were done under the supervision of the late Professor Yehuda Haas from the Physical Chemistry Department at the same university. He has been working in the Department of Chemistry Nuclear Research Center Negev (NRCN) since 1995; served as the Head of Department of Chemistry (2010-2013) at the same center. From 2013-2018 he became the Head of Nuclear Engineering Department of the Israel Atomic Energy Commission. Currently he is in sabbatical in the Department of Chemical Engineering at Ben Gurion University of the Negev (Israel).
Abstract:
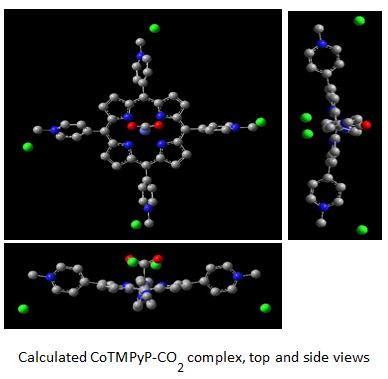
The electrochemical reduction of CO2 offers one of the possible solutions to current energy and sustainability issues since it can sequester carbon from the atmosphere and can be used to produce fuels and useful chemicals. In this respect, some metalloporphyrins have been reported to catalyze the electroreduction of CO2. However, key issues still remain in regard to the elucidation of the effect of the porphyrin structure on the reaction mechanism and catalyst activity. An essential and necessary stage in the proposed mechanism for the catalytic reduction of CO2 by the Co(II)/Co(I) porphyrin redox couple is the formation of an intermediate Co(II)porphyrin-CO2- complex. In an attempt to examine the effect of positively and negatively charged porphyrin substituents on the catalytic activity, we report here on a combined DFT and empirical study regarding the electrochemical reduction of CO2 in the presence of the Cobalt(II) 5,10,15,20-(tetra-N-methyl-4-pyridyl) porphyrin - Co(II) TMPyP and Cobalt(II) 5,10,15,20-(tetra-4-sulfonatophenyl) porphyrin – Co(II)TPPS complexes, with charges of +4 and -4, respectively. The lower catalytic activity of the CoTPPS complex as compared to that of CoTMPyP, both dissolved in aqueous alkaline solutions, as demonstrated by cyclic voltammetry experiments, are in agreement with the DFT study. Coulombic interactions seem to dictate the cobalt-carbon bond length and strength in the porphyrin-CO2 intermediate, and consequently have an impact on its stability and on the overall catalytic activity towards CO2 reduction.
Juan Manuel Lázaro-MartÃnez
University of Buenos Aires, Argentina
Title: Coordination chemistry in pyridine and imidazole compounds containing gem-diol moieties. Solid-state NMR and X-Ray studies
Time : 14:45-15:10

Biography:
Juan M Lazaro Martinez obtained his PhD in Organic Chemistry from University of Buenos Aires (UBA) in 2011. His work is based on the development, characterization and applications of hydrogel materials has received two important mentions: “Dr. Luis Federico Leloir 2012 Prize” (UBA), “SAIQO 2013 Prize” to the best PhD thesis in the area of organic synthesis (SAIQO -Argentine Society of Research in Organic Chemistry). He was a Postdoctoral fellow at the Enrique Gaviola Institute of Physics of National University of (UNC), Argentina. Currently, he is an Assistant Professor of Organic Chemistry at UBA. He is a Member of the Institute of IQUIMEFA-CONICET as a Researcher. His areas of research are focused on the synthesis and crystalline properties of poly(ethylenimine) polymers with biotechnology applications and structural elucidation in Cu- and Co-complexes studied by solid-state NMR and single-crystal X-ray techniques with environmental and synthetic applications.
Abstract:
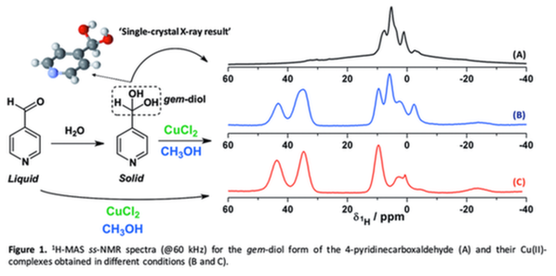
The need for clean technology either for fine chemistry or for waste treatment leads to the replacement of traditional inorganic oxidants such as K2Cr2O7 and KMnO4 by benign, easy-to-handle oxidants, such as H2O2 and O2. The activation of H2O2 can be achieved by transition-metal ion complexes with organic ligands. Particularly, H2O2 can produce OH• via the Cu(II)/Cu(I) cycle involving different reaction pathways. A high number of metal complexes bearing gem-diols has been reported, in which the presence of these moieties is generally demonstrated by single-crystal X-ray diffraction. As a rule, the stability of this functional group is not studied in the free ligand before the preparation of the metal complex. Understanding the chemistry of gem-diols is crucial for the development of synthetic methods to obtain new organic ligands, which are often used for the design of metal complexes with catalytic activity. In this context, solid-state NMR (ss-NMR) is a useful methodology to elucidate the chemical composition of mixtures in which both gem-diol and carbonyl forms are present in cases where the single-crystal cannot be obtained for X-ray studies. Additionally, the 1H-MAS ss-NMR spectra (@60 kHz) can also bring structural information about the ligands surrounding the paramagnetic center. To have an insight into the chemistry of gem-diol compounds, the aim of this work is to study the gem-diol generation and copper coordination properties in imidazole- and pyridinecarboxaldehydes through the combination of ss-NMR and single-crystal X-ray diffraction techniques. Complementary analyses were performed by solution-state NMR, high-resolution mass spectrometry (HRMS), and 1H ss- NMR. These studies allow us to expand the chemistry in metal complexes in terms of structural diversity of the ligands at the same time that new Cu(II)-homogenous catalyst towards the activation of H2O2 will be explored.
Germain Vallverdu
University of Pau & Pays Adour, France
Title: Surface reactivity of layered manganese oxides: an experimental and theoretical approach
Time : 15:10-15:35

Biography:
Germain Vallverdu is associated professor in the university of Pau & Pays Adour, France at the IPREM institute (Institute of analytical sciences and physicochemistry for environment and materials), CNRS / UPPA UMR 5254. He is a specialist in theoretical chemistry and numerical simulations. His research activities concern the development of new methods and computational strategies at different time or space scales (from quantum to classical approaches), applied to the investigations of complex systems. The computational approaches are lead in close interaction with experimentalists and take advantage of the high level instrumental platform of the institute. Examples of these systems and applications are the investigation of the chemical reactivity and the electronic properties at the surfaces and interfaces of materials used in lithium-ion cells.
Abstract:
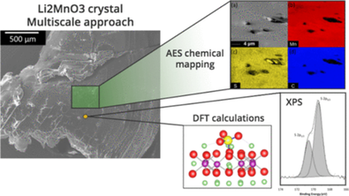
Electrochemical storage of energy through Li-ion devices is the commonly used solution to address the intermittent character of renewable energy and the increasing demand of nomad technologies. LiCoO2 is the most widely used positive electrode material of today’s Li-ion batteries. In the last decade, much research has been performed to explore alternative materials as mixed transition metal oxides LiNixMnxCo1−2xO2 (NMC). The surface reactivity of these electrode materials towards the electrolyte is a key feature that has a deep impact on the performance and lifetime of Li-ion cells and needs to be understood and controls. Within this framework, based on our previous experience on lithium layered oxides, we decided to study the surface reactivity of Li2MnO3 which can be view as a model compound for MnIV layered oxides such as NMC or even Li-rich materials. The strategy consists in coupling adsorption of gaseous probe molecule (SO2), X-ray photoelectron spectroscopy (XPS) and DFT calculation in order to identify the influence of the oxidation state of the transition metal on the adsorption reaction type (basic/acidic or redox). We focus our study on strengthening the experimental/calculation coupling by studying the reactivity on a single crystal surface of Li2MnO3. Both approaches conclude to a redox adsorption mode with the formation of sulphate species. Chemical maps of the crystal surface after adsorption obtained by Auger spectroscopy provide information on the adsorption sites location. Stacking faults and spinel type default are usually encountered in the Li2MnO3 crystals. Thus, we completed this study with the investigation of the surface reactivity of Li2MnO3 polycrystals against the stacking faults rate. Moreover, the reactivity of Li1+xMn2-xO4 spinel materials will be checked to determine the influence of the spinel type default on the surface reactivity.
Imad Ali Abu-Yousef
American University of Sharjah, UAE
Title: Synthesis and cyclic voltammetry study of asymmetrical triphenylmethane di- and trisulfides on coated and bare gold electrodes
Time : 15:50-16:15

Biography:
Imad A Abu Yousef earned a PhD in Organic Chemistry from McGill University, Canada. He pursued a Post-doctoral Fellowship in Polymer Chemistry at the same university. Prior to joining as Professor in American University of Sharjah, UAE he served as the faculty of several universities. His main research interests include sulfur chemistry, organic electrochemistry, polymer chemistry, photocatalysis, medicinal chemistry and synthesis of new organic polychalcogenides.
Abstract:
A new series of asymmetrical trityl di- (TrSSR) and trisulfides (TrSSSR) were prepared and characterized using 1H-, 13C-NMR and elemental analysis as well as by X-ray crystallography. The materials were tested for their electrochemical reduction processes using cyclic voltammetry on both bare and coated gold electrodes. On a coated gold electrode surface adsorption phenomena are absent; the electrochemical reduction process is irreversible and found to be diffusion controlled. In contrast, on a bare gold electrode surface, a self-assembled monolayer of TrSAuads and RSAuads for disulfides and TrSSAuads or RSSAuads for trisulfides originates after S-S bond breakage observed by simply dipping the gold electrode in a solution of sulfides. The data presented herein indicate that the electrochemical electron transfer process for both systems highly depends on the structure and the type of substituents where the electron transfer process causes the decomposition of the S-S bond on the bare gold electrode and the Tr-S σ bond on the coated gold electrode. The experimental approach allows estimating the values of Eored and the intrinsic barrier for the formation of the radical anions. The results were then compared with those obtained previously on a glassy carbon surface. The structures of the tested compounds are illustrated in the figure below.
Giovanna D’Angelo
University of Messina, Italy
Title: Multiple interacting collective modes and phonon gap in phospholipid membranes
Time : 16:15-16:40

Biography:
Giovanna D Angelo is currently an Associate Professor of Physics at the University of Messina (Italy). She was graduated in Physics from the University of Messina, in 1988 and was awarded PhD in Physics from the University of Messina, Italy in the year 1993. She has been working on different scientific topics in solid state physics and biophysics. Systems investigated: Glasses, Polymers, Biopolymers, Membrane pore forming peptides, Phospholipids membranes, Proteins, Hydration water in biological systems, Hydrogels.
Abstract:
Statement of the Problem: It has been widely accepted that fast sub-picosecond timescale coherent fluctuations in phospholipid membranes play a crucial role in passive transport of small molecules, a process that is fundamental for cellular metabolism. Despite this fast collective dynamics has been studied for more than a decade, the picture of these vibrational motions, involving nanometer-sized regions of the lipid membrane, is still fragmentary.
Methodology & Theoretical Orientation: In this work we show the results of an experimental investigation performed by advanced Brillouin neutron scattering, the data of which have been combined with recent inelastic X-ray scattering by Zhernenkov et al.
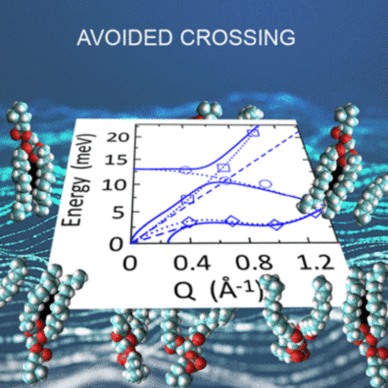
Findings: With our analysis we overcome the restrictions linked to the accessible dynamic range and the shape of the energy resolution of both the techniques. Most importantly, by interpreting the spectra with an extension of an interacting modes model, previously proposed by some of the present authors, we provide evidence for a complex scenario for the low energy collective vibrations in phospholipid bilayers, where multiple low energy optical modes exist, that play a crucial role in avoided crossing of the dispersion relations of phospholipids, as previously predicted by our MD simulation.
Conclusion & Significance: This approach allows for a comprehensive and unprecedented picture of the vibrational collective features of phospholipids. At low wavevectors Q, the dispersion relations can be interpreted in terms of two acoustic-like modes, one longitudinal and one transverse, plus a dispersion less optic-like mode. The transverse mode of the liquid phase shows a phonon gap that can be linked to a passive transport mechanism through membranes.
Caterina Branca
University of Messina, Italy
Title: Dynamical and structural characterization of thermally responsive pluronic-based nano-delivery systems
Time : 16:40-17:05

Biography:
Caterina Branca is currently an Associate Professor of Physics at the University of Messina (Italy). She completed diploma of scientific maturity at the liceo scientifico "archimede", messina, Italy in 1989 and degree in physics at the physics department of the university of messina, Italy in 1996. She was awarded PhD in physics from University of Messina in the year 2000. Her research activity concerns essentially with the study of the structural and dynamical properties of soft condensed matter, such as disaccharide aqueous solutions, polymeric systems, gels, glasses, etc. For such kind of studies a wide class of techniques has been employed such as light (Raman) and neutron scattering (QENS, INS, SANS), photon correlation spectroscopy, Fourier Transform Infrared Spectroscopy, etc. Currently, the research activity concerns with the synthesis and characterization of “smart” polymeric hydrogels and block copolymer micelles as novel carrier systems in the field of drug targeting. She is the author of about 90 articles published in international peer-reviewed journals in addition to numerous conference proceedings.
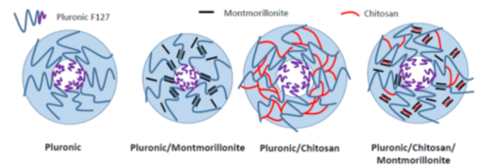
Abstract:
Statement of the Problem: The combination of pluronic and nanoparticles is currently receiving considerable interest in biomedical applications. In this context, investigating and understanding the structural and dynamical properties of pluronicbased systems is crucial to optimize the formulation of high performance multifunctional structures. So far, the effects of polysaccharides and of clays singularly added on a pluronic water dispersion were investigated. The combined addition of chitosan and montmorillonite opens the possibility to join the properties of the single constituents to formulate bio-based temperature-sensitive vehicles.
Methodology & Theoretical Orientation: Chitosan, montmorillonite and chitosan-montmorillonite nanocomposites were added on a concentrated pluronic F127 aqueous solution. The pluronic-based systems were investigated by differential scanning calorimetry (DSC), X-ray diffraction (XRD), rheology, Fourier transform infrared attenuated total reflection spectroscopy (FTIR-ATR) and dynamic light scattering (DLS). The gelation and micellization behaviors of pluronic were compared to those of the pluronic-based composites and analyzed in terms of the different elasticity of the investigated samples. FTIRATR spectroscopy was applied to analyze different vibrational modes in order to evidence differences in the conformational arrangements of the micelles. Finally, by DLS the dynamics of the pluronic-based/water systems was analyzed and depending upon solution temperature the observed decays were attributed to differently sized diffusive entities.
Conclusion & Significance: The experimental findings give strong evidence for the coexistence of complex states of aggregation allowing us to get a better insight into the architecture of the investigated systems.
Silvia Carlotto
University of Padova, Italy
Title: Transition metal systems: a theoretical modeling of their L2,3-edge X-ray absorption spectra
Time : 17:05-17:30

Biography:
Silvia Carlotto is a Researcher at the University of Padua, Italy. She started her PhD research activity in the Theoretical Chemistry Group at the same university with a thesis devoted to the modeling of dynamic solvation effects. After her PhD, she participated as Postdoc in several national and international projects about the simulation of non-linear optical properties of multipolar chromophores in solution; the development of computational methods for micro fluidic devices and the theoretical study of the next generation, cost efficient, automotive catalysis in particular and the modeling of the catalytic properties of pure and doped perovskites. During the past years, she has worked on the simulation of X-ray absorption and photoemission spectra (K- and L-edges) of isolated and supported (Fe and FeO2- phthalocyanine) systems to gain insights into their occupied and unoccupied electronic structures.
Abstract:
X-ray absorption spectroscopy (XAS) of transition metal (TM) complexes is recognized as a tool able to probe, siteselectively, the empty frontier MOs, the TM coordinative environment, the ligand -field splitting, the oxidation states and, in general the nature and the strength of the TM–ligand bonding in TM complexes. Despite metal K and L 2,3-edges XA spectra contain a huge chemical information, a stiff theoretical analysis is needed to extract it. The L 2, 3-edges XA simulated spectra herein presented have been obtained by using the restricted open shell configuration interaction with singles method, which includes spin orbit coupling and relativistic effects by employing the ORCA program package. Numerical experiments have been carried out to investigate the TM L 2, 3-edges spectra of a huge number of systems that spread between single molecules (TM (acac)2 (TM = Mn, Co) , TM (acac)3 (TM = Cr, Mn, Fe), VPc and VXPc systems (X = O, I) Iron complexes (scorpionate, ferrocene and bridged carbonyl ones) and 2D complex systems (Ag supported FePc/FePc(η2-O2) and Cu supported THQ/ THQ:Cu4). Instead, K-edge spectra have been modeled by running time dependent density functional theory calculations and by using the ADF program package. Numerical experiments on the O, C, F, N K-edges have been performed on TM(acac)3 and CuPc/CuTPP systems. The goals of these systematic studies is: i) to reveal the role played by the TM, by its oxidation state and its environment in determining the spectral features, ii) to provide an intimate understanding of the electron transfer pathway ruling the catalytic oxygen reduction reaction of FePc on Ag (110) and iii) to quantify the amount of different species that contribute to the same XA spectrum. Relevant trends for L3-edge XA spectra are the lower energy side which is characterized by TM→TM transitions, while the higher energy one involves metal to ligand charge transfer transitions.
Sharipov Rustam Hasanovich
Kazakh-British Technical University, Republic of Kazakhstan
Title: Application of combined electrochemical reactions for extraction of metals from metal- containing waste
Time : 17:30-17:45

Biography:
Sharipov Rustam Hasanovich has successfully completed his graduation in 2005. Currently he is pursuing PhD majoring in metallurgy from the Kazakh-British Technical University, Republic of Kazakhstan. He studied at the Kazakh National Technical University named after K I Satpayev specializing in metallurgy (2005-2009); also specialized in materials science and technology of new materials from the same university (2010 to 2012). He worked in the Laboratory of Technology of Electrochemical Productions as an Engineer Researcher at the National Center for Complex Processing of Mineral Raw Materials, Republic of Kazakhstan (2009-2013).
Abstract:
The method of application of combined electrochemical reactions is designed for the purpose of obtaining a leaching agent and extraction of metals into a solution from various raw materials in the volume of one reactor. In our experiments, the initial solution was a solution of NaOH. As a source of sulfur for the production of leaching reagents, a sulfur-graphite electrode was used. A study of the electrochemical leaching of metals was carried out in a thermostatic reaction cell. In this case, a sulfur-graphite electrode (SGE) was used as a cathode, and graphite served as an anode. When metals were extracted into the solution, it was shown that practically all physico-chemical factors influence the leaching process, and the microstructure of inorganic aqueous solutions should be considered as one of the important parameters of the technological process. It is shown that the characteristics of solutions vary not only from the change in the concentration of dissolved substances, but also when all the physico-chemical conditions for the leaching of metal-containing raw materials change. We carried out experiments on the transfer into the solution of metals from alloys 1: chemical composition %: Pb - 47.52, Bi - 46.31, Cd - 6.17; from alloy 2 chemical composition %: Cd - 18.83, Ti - 16.93, Ag - 15.49, Sn - 13.28, V - 6.07, Co - 0.11, Ni - 2.18 , Cu - 3.54, Mn - 3.71, Fe - 5.79, Pb - 1.19 and from brass: chemical composition %: Cu - 58.65, Zn - 39.79, Pb - 1.34, Cr - 0.06, N i - 0.05, Nb - 0.11. For example, the degree of silver recovery during electroleaching for 6 hours increases with a NaOH concentration of 0.2 M to 8.2%, 0.5 M to 8.2%, 1.0 M to 12.2%, 2.0 M to 12.6%. For tin, 0.2 M is 6.9%, 0.5 M is 7.6%, 1.0 M is 7.8%, 2.0 M is 8.0%. To achieve the optimum extraction value, studies are continuing to determine the structure of compounds in metallic alloys. Studies have shown that, with optimal technical design, the method of combined electrochemical reactions can be applied to any type of metal-containing raw material.
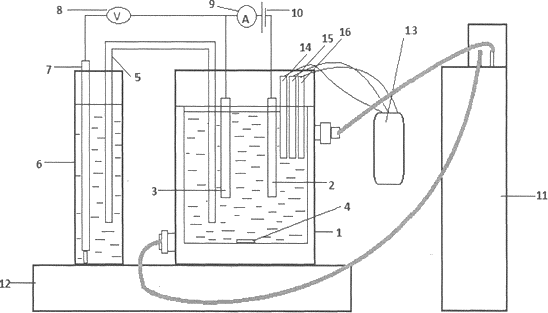
Figure 1: Schematic of an electrochemical cell for leaching.
1 - cell; 2 - graphite electrode; 3 - sulfur/graphite electrode; 4 - a magnet for magnetic stirrer; 5 - a glass bridge; 6 - glass with sodium hydroxide; 7 -silver chloride electrode; 8 - universal voltmeter; 9 - Amperemeter; 10 - power supply; 11 - thermostat; 12 - magnetic stirrer; 13 - multiparameter measuring device (Sens Ion 156); 14 - an electrode for measuring the pH of the medium; 15 - electrode for measuring the concentration of dissolved oxygen; 16 - electrode for measuring the electrical conductivity.
Myrzakhanov Maxat Makhmudovich
Kazakh-British Technical University, Republic of Kazakhstan
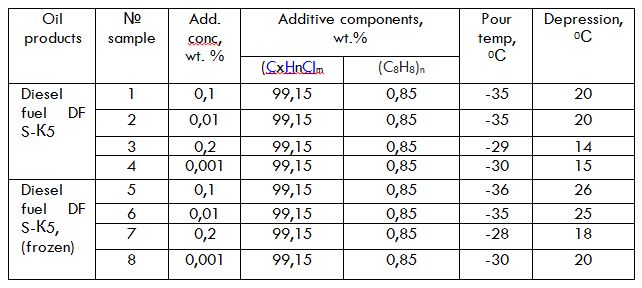
Title: Composition for reducing the pour point of diesel fuel
Time : 17:45-18:00

Biography:
Myrzakhanov MM, born in 1990, graduated from Kazakhstan-British Technical University in 2012 with a degree in chemical technology of organic substances. In 2014, he graduated from the Kazakh-British Technical University with a degree in Petrochemistry. In 2016, he entered a doctorate in the specialty "Petrochemistry" in the Kazakh National Research Technical University named after K. Satpaev. Since 2012 he has been working at JSC KBTU, in the research laboratory "Advanced Materials and Technologies" as a researcher. Myrzakhanov M.M. is the executor of the research topic on the project "Development of catalytic systems and technology of conversion of natural gas into dimethyl ether" for 2015-2017.
Abstract:
The current economic situation in the country and the high physical wear and tear on the equipment of domestic refineries make it possible to influence the quality of diesel fuel only by reducing the beginning of its boiling. It should be noted that less than 1% of the produced diesel fuel is produced from the diesel fuel and about 10% is winter, the rest is summer, which does not correspond to the climate of our country at all. Forced use of summer diesel fuel in winter conditions leads to a huge over expenditure. At present, the production of high-quality diesel fuels is impossible without the addition of additives of various functional purposes, such as cetane-raising, anti-wear, anti-smoke, detergent, antioxidant, dispersant, depressant and others.
In developing depressant additives to diesel fuel a lot of work has been done. Many compositions of popular solvents and additives were studied, as a result of which we came to the final choice of the necessary components for the development of this additive. Numerous organic solvents were used as the solvent, but for this type of additive, it did not show satisfactory results, which led to a study of the characteristics of the desired solvents with suitable properties.
The main component was polystyrene. As we all know, polystyrene has a low density (1060 kg/m³), has excellent dielectric properties and good frost resistance (down to -40 °C), it suited as an excellent solvent for obtaining a depressant additive. It also has a low chemical resistance (in addition to dilute acids, alcohols and alkalis). It dissolves in acetone, toluene, dichloroethane, more slowly in gasoline.
As a result of the tests, excellent results were obtained, but with varying concentrations, the results varied. Therefore, the task was to determine the optimum concentrations of the components in the depressant additive. After carrying out numerous tests with varying concentrations of components, data were obtained that allowed to determine those values at which we obtained the most rational values.
To improve the low-temperature properties of hydrotreated diesel fuel, we first proposed the use of additives based on polymer ethyl derivatives of benzene. It was found that the depressant additive is a 10% polymer solution in a chlorinated alkane. The introduction of an additive based on PC-10 in an amount of 100-1500 ppm in hydrotreated diesel fuel reduces the pour point of the hydrotreated diesel fuel (Table 1). When comparing the pour point of diesel fuel with the Dodiflow commercial additive, it is established that the action of the additive based on PC-10 is more effective than in the case of commercial additive, which is a polymer of ethyl derivatives of benzene. The pour point of the diesel fuel with the additive PC-10 is -35 °Ð¡, whereas when using the commercial additive -34 °Ð¡.
The additive developed on the basis of the polymer of ethyl benzene derivatives is not inferior in effectiveness to the currently used commercial additive Dodiflow.
Table 1. Temperature of solidification of diesel fuel with depressant additives
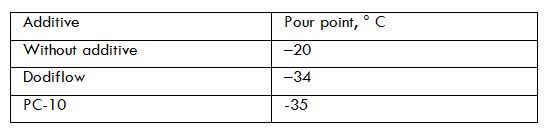
Table 2. Changes in the pour point in diesel fuel